Electron-Electron Interactions(Pdf)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Competition Between Paramagnetism and Diamagnetism in Charged
Competition between paramagnetism and diamagnetism in charged Fermi gases Xiaoling Jian, Jihong Qin, and Qiang Gu∗ Department of Physics, University of Science and Technology Beijing, Beijing 100083, China (Dated: November 10, 2018) The charged Fermi gas with a small Lande-factor g is expected to be diamagnetic, while that with a larger g could be paramagnetic. We calculate the critical value of the g-factor which separates the dia- and para-magnetic regions. In the weak-field limit, gc has the same value both at high and low temperatures, gc = 1/√12. Nevertheless, gc increases with the temperature reducing in finite magnetic fields. We also compare the gc value of Fermi gases with those of Boltzmann and Bose gases, supposing the particle has three Zeeman levels σ = 1, 0, and find that gc of Bose and Fermi gases is larger and smaller than that of Boltzmann gases,± respectively. PACS numbers: 05.30.Fk, 51.60.+a, 75.10.Lp, 75.20.-g I. INTRODUCTION mK and 10 µK, respectively. The ions can be regarded as charged Bose or Fermi gases. Once the quantum gas Magnetism of electron gases has been considerably has both the spin and charge degrees of freedom, there studied in condensed matter physics. In magnetic field, arises the competition between paramagnetism and dia- the magnetization of a free electron gas consists of two magnetism, as in electrons. Different from electrons, the independent parts. The spin magnetic moment of elec- g-factor for different magnetic ions is diverse, ranging trons results in the paramagnetic part (the Pauli para- from 0 to 2. -

Lecture 3: Fermi-Liquid Theory 1 General Considerations Concerning Condensed Matter
Phys 769 Selected Topics in Condensed Matter Physics Summer 2010 Lecture 3: Fermi-liquid theory Lecturer: Anthony J. Leggett TA: Bill Coish 1 General considerations concerning condensed matter (NB: Ultracold atomic gasses need separate discussion) Assume for simplicity a single atomic species. Then we have a collection of N (typically 1023) nuclei (denoted α,β,...) and (usually) ZN electrons (denoted i,j,...) interacting ∼ via a Hamiltonian Hˆ . To a first approximation, Hˆ is the nonrelativistic limit of the full Dirac Hamiltonian, namely1 ~2 ~2 1 e2 1 Hˆ = 2 2 + NR −2m ∇i − 2M ∇α 2 4πǫ r r α 0 i j Xi X Xij | − | 1 (Ze)2 1 1 Ze2 1 + . (1) 2 4πǫ0 Rα Rβ − 2 4πǫ0 ri Rα Xαβ | − | Xiα | − | For an isolated atom, the relevant energy scale is the Rydberg (R) – Z2R. In addition, there are some relativistic effects which may need to be considered. Most important is the spin-orbit interaction: µ Hˆ = B σ (v V (r )) (2) SO − c2 i · i × ∇ i Xi (µB is the Bohr magneton, vi is the velocity, and V (ri) is the electrostatic potential at 2 3 2 ri as obtained from HˆNR). In an isolated atom this term is o(α R) for H and o(Z α R) for a heavy atom (inner-shell electrons) (produces fine structure). The (electron-electron) magnetic dipole interaction is of the same order as HˆSO. The (electron-nucleus) hyperfine interaction is down relative to Hˆ by a factor µ /µ 10−3, and the nuclear dipole-dipole SO n B ∼ interaction by a factor (µ /µ )2 10−6. -

Solid State Physics 2 Lecture 5: Electron Liquid
Physics 7450: Solid State Physics 2 Lecture 5: Electron liquid Leo Radzihovsky (Dated: 10 March, 2015) Abstract In these lectures, we will study itinerate electron liquid, namely metals. We will begin by re- viewing properties of noninteracting electron gas, developing its Greens functions, analyzing its thermodynamics, Pauli paramagnetism and Landau diamagnetism. We will recall how its thermo- dynamics is qualitatively distinct from that of a Boltzmann and Bose gases. As emphasized by Sommerfeld (1928), these qualitative di↵erence are due to the Pauli principle of electons’ fermionic statistics. We will then include e↵ects of Coulomb interaction, treating it in Hartree and Hartree- Fock approximation, computing the ground state energy and screening. We will then study itinerate Stoner ferromagnetism as well as various response functions, such as compressibility and conduc- tivity, and screening (Thomas-Fermi, Debye). We will then discuss Landau Fermi-liquid theory, which will allow us understand why despite strong electron-electron interactions, nevertheless much of the phenomenology of a Fermi gas extends to a Fermi liquid. We will conclude with discussion of electrons on the lattice, treated within the Hubbard and t-J models and will study transition to a Mott insulator and magnetism 1 I. INTRODUCTION A. Outline electron gas ground state and excitations • thermodynamics • Pauli paramagnetism • Landau diamagnetism • Hartree-Fock theory of interactions: ground state energy • Stoner ferromagnetic instability • response functions • Landau Fermi-liquid theory • electrons on the lattice: Hubbard and t-J models • Mott insulators and magnetism • B. Background In these lectures, we will study itinerate electron liquid, namely metals. In principle a fully quantum mechanical, strongly Coulomb-interacting description is required. -

Unconventional Hund Metal in a Weak Itinerant Ferromagnet
ARTICLE https://doi.org/10.1038/s41467-020-16868-4 OPEN Unconventional Hund metal in a weak itinerant ferromagnet Xiang Chen1, Igor Krivenko 2, Matthew B. Stone 3, Alexander I. Kolesnikov 3, Thomas Wolf4, ✉ ✉ Dmitry Reznik 5, Kevin S. Bedell6, Frank Lechermann7 & Stephen D. Wilson 1 The physics of weak itinerant ferromagnets is challenging due to their small magnetic moments and the ambiguous role of local interactions governing their electronic properties, 1234567890():,; many of which violate Fermi-liquid theory. While magnetic fluctuations play an important role in the materials’ unusual electronic states, the nature of these fluctuations and the paradigms through which they arise remain debated. Here we use inelastic neutron scattering to study magnetic fluctuations in the canonical weak itinerant ferromagnet MnSi. Data reveal that short-wavelength magnons continue to propagate until a mode crossing predicted for strongly interacting quasiparticles is reached, and the local susceptibility peaks at a coher- ence energy predicted for a correlated Hund metal by first-principles many-body theory. Scattering between electrons and orbital and spin fluctuations in MnSi can be understood at the local level to generate its non-Fermi liquid character. These results provide crucial insight into the role of interorbital Hund’s exchange within the broader class of enigmatic multiband itinerant, weak ferromagnets. 1 Materials Department, University of California, Santa Barbara, CA 93106, USA. 2 Department of Physics, University of Michigan, Ann Arbor, MI 48109, USA. 3 Neutron Scattering Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA. 4 Institute for Solid State Physics, Karlsruhe Institute of Technology, 76131 Karlsruhe, Germany. -
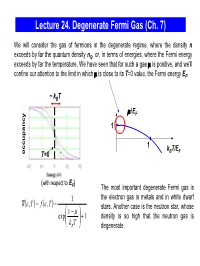
Lecture 24. Degenerate Fermi Gas (Ch
Lecture 24. Degenerate Fermi Gas (Ch. 7) We will consider the gas of fermions in the degenerate regime, where the density n exceeds by far the quantum density nQ, or, in terms of energies, where the Fermi energy exceeds by far the temperature. We have seen that for such a gas μ is positive, and we’ll confine our attention to the limit in which μ is close to its T=0 value, the Fermi energy EF. ~ kBT μ/EF 1 1 kBT/EF occupancy T=0 (with respect to E ) F The most important degenerate Fermi gas is 1 the electron gas in metals and in white dwarf nε()(),, T= f ε T = stars. Another case is the neutron star, whose ε⎛ − μ⎞ exp⎜ ⎟ +1 density is so high that the neutron gas is ⎝kB T⎠ degenerate. Degenerate Fermi Gas in Metals empty states ε We consider the mobile electrons in the conduction EF conduction band which can participate in the charge transport. The band energy is measured from the bottom of the conduction 0 band. When the metal atoms are brought together, valence their outer electrons break away and can move freely band through the solid. In good metals with the concentration ~ 1 electron/ion, the density of electrons in the electron states electron states conduction band n ~ 1 electron per (0.2 nm)3 ~ 1029 in an isolated in metal electrons/m3 . atom The electrons are prevented from escaping from the metal by the net Coulomb attraction to the positive ions; the energy required for an electron to escape (the work function) is typically a few eV. -

Chapter 13 Ideal Fermi
Chapter 13 Ideal Fermi gas The properties of an ideal Fermi gas are strongly determined by the Pauli principle. We shall consider the limit: k T µ,βµ 1, B � � which defines the degenerate Fermi gas. In this limit, the quantum mechanical nature of the system becomes especially important, and the system has little to do with the classical ideal gas. Since this chapter is devoted to fermions, we shall omit in the following the subscript ( ) that we used for the fermionic statistical quantities in the previous chapter. − 13.1 Equation of state Consider a gas ofN non-interacting fermions, e.g., electrons, whose one-particle wave- functionsϕ r(�r) are plane-waves. In this case, a complete set of quantum numbersr is given, for instance, by the three cartesian components of the wave vector �k and thez spin projectionm s of an electron: r (k , k , k , m ). ≡ x y z s Spin-independent Hamiltonians. We will consider only spin independent Hamiltonian operator of the type ˆ 3 H= �k ck† ck + d r V(r)c r†cr , �k � where thefirst and the second terms are respectively the kinetic and th potential energy. The summation over the statesr (whenever it has to be performed) can then be reduced to the summation over states with different wavevectork(p=¯hk): ... (2s + 1) ..., ⇒ r � �k where the summation over the spin quantum numberm s = s, s+1, . , s has been taken into account by the prefactor (2s + 1). − − 159 160 CHAPTER 13. IDEAL FERMI GAS Wavefunctions in a box. We as- sume that the electrons are in a vol- ume defined by a cube with sidesL x, Ly,L z and volumeV=L xLyLz. -

Landau Effective Interaction Between Quasiparticles in a Bose-Einstein Condensate
PHYSICAL REVIEW X 8, 031042 (2018) Landau Effective Interaction between Quasiparticles in a Bose-Einstein Condensate A. Camacho-Guardian* and Georg M. Bruun Department of Physics and Astronomy, Aarhus University, Ny Munkegade, DK-8000 Aarhus C, Denmark (Received 19 December 2017; revised manuscript received 28 February 2018; published 15 August 2018) Landau’s description of the excitations in a macroscopic system in terms of quasiparticles stands out as one of the highlights in quantum physics. It provides an accurate description of otherwise prohibitively complex many-body systems and has led to the development of several key technologies. In this paper, we investigate theoretically the Landau effective interaction between quasiparticles, so-called Bose polarons, formed by impurity particles immersed in a Bose-Einstein condensate (BEC). In the limit of weak interactions between the impurities and the BEC, we derive rigorous results for the effective interaction. They show that it can be strong even for a weak impurity-boson interaction, if the transferred momentum- energy between the quasiparticles is resonant with a sound mode in the BEC. We then develop a diagrammatic scheme to calculate the effective interaction for arbitrary coupling strengths, which recovers the correct weak-coupling results. Using this scheme, we show that the Landau effective interaction, in general, is significantly stronger than that between quasiparticles in a Fermi gas, mainly because a BEC is more compressible than a Fermi gas. The interaction is particularly large near the unitarity limit of the impurity-boson scattering or when the quasiparticle momentum is close to the threshold for momentum relaxation in the BEC. -

Attractive Fermi Polarons at Nonzero Temperatures with a Finite Impurity
PHYSICAL REVIEW A 98, 013626 (2018) Attractive Fermi polarons at nonzero temperatures with a finite impurity concentration Hui Hu, Brendan C. Mulkerin, Jia Wang, and Xia-Ji Liu Centre for Quantum and Optical Science, Swinburne University of Technology, Melbourne, Victoria 3122, Australia (Received 29 June 2018; published 25 July 2018) We theoretically investigate how quasiparticle properties of an attractive Fermi polaron are affected by nonzero temperature and finite impurity concentration in three dimensions and in free space. By applying both non- self-consistent and self-consistent many-body T -matrix theories, we calculate the polaron energy (including decay rate), effective mass, and residue, as functions of temperature and impurity concentration. The temperature and concentration dependencies are weak on the BCS side with a negative impurity-medium scattering length. Toward the strong attraction regime across the unitary limit, we find sizable dependencies. In particular, with increasing temperature the effective mass quickly approaches the bare mass and the residue is significantly enhanced. At temperature T ∼ 0.1TF ,whereTF is the Fermi temperature of the background Fermi sea, the residual polaron-polaron interaction seems to become attractive. This leads to a notable down-shift in the polaron energy. We show that, by taking into account the temperature and impurity concentration effects, the measured polaron energy in the first Fermi polaron experiment [Schirotzek et al., Phys.Rev.Lett.102, 230402 (2009)] could be better theoretically explained. DOI: 10.1103/PhysRevA.98.013626 I. INTRODUCTION Experimentally, the first experiment on attractive Fermi polarons was carried out by the Zwierlein group at Mas- Over the past two decades, ultracold atomic gases have pro- sachusetts Institute of Technology (MIT) in 2009 using 6Li vided an ideal platform to understand the intriguing quantum many-body systems [1]. -

Schedule for Rest of Quarter • Review: 3D Free Electron Gas • Heat Capacity
Lecture 16 Housekeeping: schedule for rest of quarter Review: 3D free electron gas Heat capacity of free electron gas Semi classical treatment of electric conductivity Review: 3D fee electron gas This model ignores the lattice and only treats the electrons in a metal. It assumes that electrons do not interact with each other except for pauli exclusion. 풌∙풓 Wavelike-electrons defined by their momentum eigenstate k: 휓풌(풓) = 푒 Treat electrons like quantum particle in a box: confined to a 3D box of dimensions L in every direction Periodic boundary conditions on wavefunction: 휓(푥 + 퐿, 푦, 푧) = 휓(푥, 푦, 푧) 휓(푥, 푦 + 퐿, 푧) = 휓(푥, 푦, 푧) 휓(푥, 푦, 푧 + 퐿) = 휓(푥, 푦, 푧) Permissible values of k of the form: 2휋 4휋 푘 = 0, ± , ± , … 푥 퐿 퐿 Free electron schrodinger’s eqn in 3D + boundary conditions gives energy eigenvalues: ℏ2 휕2 휕2 휕2 − ( + + ) 휓 (푟) = 휖 휓 (푟) 2푚 휕푥2 휕푦2 휕푧2 푘 푘 푘 ℏ2 ℏ2푘2 (푘2 + 푘2 + 푘2) = 휖 = 2푚 푥 푦 푧 푘 2푚 There are N electrons and we put them into available eigenstates following these rules: o Every state is defined by a unique quantized value of (푘푥, 푘푦, 푘푧) o Every state can hold one spin up and one spin down electron o Assume that single-electron solution above applies to collection of electrons (i.e. electrons do not interact and change the eigenstates) o Fill low energy states first. In 3D, this corresponds to filling up a sphere in k space, one 2 2 2 2 ‘shell’ at a time. -

Chapter 6 Free Electron Fermi
Chapter 6 Free Electron Fermi Gas Free electron model: • The valence electrons of the constituent atoms become conduction electrons and move about freely through the volume of the metal. • The simplest metals are the alkali metals– lithium, sodium, potassium, Na, cesium, and rubidium. • The classical theory had several conspicuous successes, notably the derivation of the form of Ohm’s law and the relation between the electrical and thermal conductivity. • The classical theory fails to explain the heat capacity and the magnetic susceptibility of the conduction electrons. M = B • Why the electrons in a metal can move so freely without much deflections? (a) A conduction electron is not deflected by ion cores arranged on a periodic lattice, because matter waves propagate freely in a periodic structure. (b) A conduction electron is scattered only infrequently by other conduction electrons. Pauli exclusion principle. Free Electron Fermi Gas: a gas of free electrons subject to the Pauli Principle ELECTRON GAS MODEL IN METALS Valence electrons form the electron gas eZa -e(Za-Z) -eZ Figure 1.1 (a) Schematic picture of an isolated atom (not to scale). (b) In a metal the nucleus and ion core retain their configuration in the free atom, but the valence electrons leave the atom to form the electron gas. 3.66A 0.98A Na : simple metal In a sea of conduction of electrons Core ~ occupy about 15% in total volume of crystal Classical Theory (Drude Model) Drude Model, 1900AD, after Thompson’s discovery of electrons in 1897 Based on the concept of kinetic theory of neutral dilute ideal gas Apply to the dense electrons in metals by the free electron gas picture Classical Statistical Mechanics: Boltzmann Maxwell Distribution The number of electrons per unit volume with velocity in the range du about u 3/2 2 fB(u) = n (m/ 2pkBT) exp (-mu /2kBT) Success: Failure: (1) The Ohm’s Law , (1) Heat capacity Cv~ 3/2 NKB the electrical conductivity The observed heat capacity is only 0.01, J = E , = n e2 / m, too small. -

Fermi and Bose Gases
Fermi and Bose gases Anders Malthe-Sørenssen 25. november 2013 1 1 Thermodynamic identities and Legendre transforms 2 Fermi-Dirac and Bose-Einstein distribution When we discussed the ideal gas we assumed that quantum effects were not important. This was implied when we introduced the term 1=N! for the partition function for the ideal gas, because this term only was valid in the limit when the number of states are many compared to the number of particles. Now, we will address the general case. We start from an individual particle. For a single particle, we address its orbitals: Its possible quantum states, each of which corresponds to a solution of the Schrodinger equation for a single particle. For many particles we will address how the “fill up" the various orbitals { where the underlying assump- tion is that we can treat a many-particle system as a system with the same energy levels as for a single particular system, that is as a system with no interactions between the states of the various particles. However, the number of particles that can occupy the same state depends on the type of particle: For Fermions (1/2 spin) Only 0 or 1 fermion can be in a particular state. For Bosons (integer spin) Any number of bosons can be in a particular state. We will now apply a Gibbs ensemble approach to study this system. This means that we will consider a system where µ, V; T is constant. We can use the Gibbs sum formalism to address the behavior of this system. -

Non-Fermi Liquids in Oxide Heterostructures
UC Santa Barbara UC Santa Barbara Previously Published Works Title Non-Fermi liquids in oxide heterostructures Permalink https://escholarship.org/uc/item/1cn238xw Journal Reports on Progress in Physics, 81(6) ISSN 0034-4885 1361-6633 Authors Stemmer, Susanne Allen, S James Publication Date 2018-06-01 DOI 10.1088/1361-6633/aabdfa Peer reviewed eScholarship.org Powered by the California Digital Library University of California Reports on Progress in Physics KEY ISSUES REVIEW Non-Fermi liquids in oxide heterostructures To cite this article: Susanne Stemmer and S James Allen 2018 Rep. Prog. Phys. 81 062502 View the article online for updates and enhancements. This content was downloaded from IP address 128.111.119.159 on 08/05/2018 at 17:09 IOP Reports on Progress in Physics Reports on Progress in Physics Rep. Prog. Phys. Rep. Prog. Phys. 81 (2018) 062502 (12pp) https://doi.org/10.1088/1361-6633/aabdfa 81 Key Issues Review 2018 Non-Fermi liquids in oxide heterostructures © 2018 IOP Publishing Ltd Susanne Stemmer1 and S James Allen2 RPPHAG 1 Materials Department, University of California, Santa Barbara, CA 93106-5050, United States of America 062502 2 Department of Physics, University of California, Santa Barbara, CA 93106-9530, United States of America S Stemmer and S J Allen E-mail: [email protected] Received 18 July 2017, revised 25 January 2018 Accepted for publication 13 April 2018 Published 8 May 2018 Printed in the UK Corresponding Editor Professor Piers Coleman ROP Abstract Understanding the anomalous transport properties of strongly correlated materials is one of the most formidable challenges in condensed matter physics.