Physical Organic Terminology, After Ingold
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Organic Chemistry –II
Subject Chemistry Paper No and Title Paper 5: Organic Chemistry –II Module No and Title Module 5: Methods of determining mechanisms and Isotope effects Module Tag CHE_P5_M5_e-Text CHEMISTRY PAPER No. 5: Organic Chemistry -II MODULE No. 5: Methods of determining mechanisms and Isotope effects TABLE OF CONTENTS 1. Learning Outcomes 2. Introduction 3. Methods of determining mechanism 3.1 Determination of the products formed 3.2 Study of Intermediate formed 3.3 Study of catalyst 3.4 Stereochemical Evidence 3.5 Kinetic evidence 3.6 Isotope Labelling 4. Isotopic Effects 5. Summary CHEMISTRY PAPER No. 5: Organic Chemistry -II MODULE No. 5: Methods of determining mechanisms and Isotope effects 1. Learning Outcomes After studying this module, you shall be able to • Know what do we mean by mechanism of a reaction • Learn the ways to determine mechanism of a reaction • Identify the reactants, products and intermediates involved in a reaction • Evaluate the steps involved in a reaction 2. Introduction In scientific experiments and chemical reactions, all we can do is try to account for the observations by proposing theories and mechanisms. Reaction mechanisms have been an integral part of the teaching of organic chemistry and in the planning of routes for organic syntheses for about 50 years. The first sentence of Hammett’s influential book, Physical Organic Chemistry, states, “A major part of the job of the chemist is the prediction and control of the course of chemical reactions” In a chemical reaction, mechanism depicts the actual process by which the reaction has taken place. It indicates which bonds are broken, in what order, the steps involved and the relative rate of each step. -

CHEMICAL KINETICS Pt 2 Reaction Mechanisms Reaction Mechanism
Reaction Mechanism (continued) CHEMICAL The reaction KINETICS 2 C H O +→5O + 6CO 4H O Pt 2 3 4 3 2 2 2 • has many steps in the reaction mechanism. Objectives ! Be able to describe the collision and Reaction Mechanisms transition-state theories • Even though a balanced chemical equation ! Be able to use the Arrhenius theory to may give the ultimate result of a reaction, determine the activation energy for a reaction and to predict rate constants what actually happens in the reaction may take place in several steps. ! Be able to relate the molecularity of the reaction and the reaction rate and • This “pathway” the reaction takes is referred to describle the concept of the “rate- as the reaction mechanism. determining” step • The individual steps in the larger overall reaction are referred to as elementary ! Be able to describe the role of a catalyst and homogeneous, heterogeneous and reactions. enzyme catalysis Reaction Mechanisms Often Used Terms •Intermediate: formed in one step and used up in a subsequent step and so is never seen as a product. The series of steps by which a chemical reaction occurs. •Molecularity: the number of species that must collide to produce the reaction indicated by that A chemical equation does not tell us how step. reactants become products - it is a summary of the overall process. •Elementary Step: A reaction whose rate law can be written from its molecularity. •uni, bi and termolecular 1 Elementary Reactions Elementary Reactions • Consider the reaction of nitrogen dioxide with • Each step is a singular molecular event carbon monoxide. -

Elimination Reactions Are Described
Introduction In this module, different types of elimination reactions are described. From a practical standpoint, elimination reactions widely used for the generation of double and triple bonds in compounds from a saturated precursor molecule. The presence of a good leaving group is a prerequisite in most elimination reactions. Traditional classification of elimination reactions, in terms of the molecularity of the reaction is employed. How the changes in the nature of the substrate as well as reaction conditions affect the mechanism of elimination are subsequently discussed. The stereochemical requirements for elimination in a given substrate and its consequence in the product stereochemistry is emphasized. ELIMINATION REACTIONS Objective and Outline beta-eliminations E1, E2 and E1cB mechanisms Stereochemical considerations of these reactions Examples of E1, E2 and E1cB reactions Alpha eliminations and generation of carbene I. Basics Elimination reactions involve the loss of fragments or groups from a molecule to generate multiple bonds. A generalized equation is shown below for 1,2-elimination wherein the X and Y from two adjacent carbon atoms are removed, elimination C C C C -XY X Y Three major types of elimination reactions are: α-elimination: two atoms or groups are removed from the same atom. It is also known as 1,1-elimination. H R R C X C + HX R Both H and X are removed from carbon atom here R Carbene β-elimination: loss of atoms or groups on adjacent atoms. It is also H H known as 1,2- elimination. R C C R R HC CH R X H γ-elimination: loss of atoms or groups from the 1st and 3rd positions as shown below. -

FULL PAPER a Self-Assembled Cage with Endohedral Acid Groups Both
FULL PAPER A Self-Assembled Cage with Endohedral Acid Groups both Catalyzes Substitution Reactions and Controls their Molecularity Paul M. Bogie, Lauren R. Holloway, Courtney Ngai, Tabitha F. Miller, Divine K. Grewal, and Richard J. Hooley[a]* [16],[17] Abstract: A self-assembled Fe4L6 cage complex internally decorated many possibilities in controlled biomimetic catalysis, above with acid functions is capable of accelerating the thioetherification of and beyond simply increasing the effective concentration of activated alcohols, ethers and amines by up to 1000-fold. No product bound substrate. The incorporation of active functions in an inhibition is seen, and effective supramolecular catalysis can occur enclosed space enables reagent-controlled reactions to take with as little as 5 % cage. The substrates are bound in the host with place in enclosed cavities, as opposed to cycloadditions[18]-[20] or up to micromolar affinities, whereas the products show binding that is unimolecular rearrangements, [21],[22] which are still the most an order of magnitude weaker. Most importantly, the cage host alters common reactions studied in synthetic hosts. By internalizing the molecularity of the reaction: whereas the reaction catalyzed by reactive functional groups in a cage, the effect of substrate simple acids is a unimolecular, SN1-type substitution process, the rate binding on nucleophilic substitution reactions can be investigated. of the host-mediated process is dependent on the concentration of nucleophile. The molecularity of the cage-catalyzed reaction is substrate-dependent, and can be up to bimolecular. In addition, the catalysis can be prevented by a large excess of nucleophile, where substrate inhibition dominates, and the use of tritylated anilines as substrates causes a negative feedback loop, whereby the liberated product destroys the catalyst and stops the reaction. -

DEPARTMENT of CHEMISTRY for M.Sc in Chemistry
DEPARTMENT OF CHEMISTRY SCHEME OF INSTRUCTIONS AND SYLLABUS (Course Book) FOR M.Sc in Chemistry Visvesvaraya National Institute of Technology, Nagpur July 2015 1 Contents Sr. No. Title Page No. 1 General Information about the department 3 2 Brief about M.Sc program 3 3 Vision and mission 4 4 Credit requirements 5 5 Detailed scheme 6 6 Detailed syllabus 7-31 2 1. General Information about the department Science is basic foundation of any technological and engineering creation. In view of the changing scenario at national and international level in field of Science and Technology, there is great demand for basic sciences with considerable knowledge of its applications. VNIT is committed to high academic standards. The M.Sc. courses are designed for four semesters (two years) in such a way that a good basic foundation of subjects is laid and applications along with recent developments are covered. Relative grading will be followed and credits will be allotted based on academic performance. Students will also get theoretical and practical knowledge of computer programming. These M.Sc. programmes provide opportunity to make career in R&D, industries and academic institutions. Opportunity for the placement may be provided by the Institute. 2. Brief about M.Sc program: Department of Chemistry offers M.Sc. (Chemistry) program which gives good foundation of basics and research component through practical skills, which in turn will provide excellent job prospects in Academics, Industries and other field of interest. M.Sc. (Chemistry) will provide competence to tackle frontier area in Green chemistry, supramolecular chemistry, Sensors, Advanced materials and Advanced organic chemistry. -
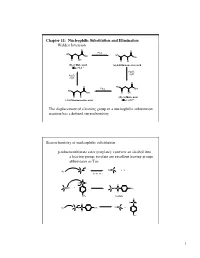
Chapter 11: Nucleophilic Substitution and Elimination Walden Inversion
Chapter 11: Nucleophilic Substitution and Elimination Walden Inversion O O PCl5 HO HO OH OH O OH O Cl (S)-(-) Malic acid (+)-2-Chlorosuccinic acid [a]D= -2.3 ° Ag2O, H2O Ag2O, H2O O O HO PCl5 OH HO OH O OH O Cl (R)-(+) Malic acid (-)-2-Chlorosuccinic acid [a]D= +2.3 ° The displacement of a leaving group in a nucleophilic substitution reaction has a defined stereochemistry Stereochemistry of nucleophilic substitution p-toluenesulfonate ester (tosylate): converts an alcohol into a leaving group; tosylate are excellent leaving groups. abbreviates as Tos C X Nu C + X- Nu: X= Cl, Br, I O Cl S O O + C OH C O S CH3 O CH3 tosylate O -O S O O Nu C + Nu: C O S CH3 O CH3 1 O Tos-Cl - H3C O O H + TosO - H O H pyridine H O Tos O CH3 [a]D= +33.0 [a]D= +31.1 [a]D= -7.06 HO- HO- O - Tos-Cl H3C O - H O O TosO + H O H pyridine Tos H O CH3 [a]D= -7.0 [a]D= -31.0 [a]D= -33.2 The nucleophilic substitution reaction “inverts” the Stereochemistry of the carbon (electrophile)- Walden inversion Kinetics of nucleophilic substitution Reaction rate: how fast (or slow) reactants are converted into product (kinetics) Reaction rates are dependent upon the concentration of the reactants. (reactions rely on molecular collisions) H H Consider: HO C _ _ C Br Br HO H H H H At a given temperature: If [OH-] is doubled, then the reaction rate may be doubled If [CH3-Br] is doubled, then the reaction rate may be doubled A linear dependence of rate on the concentration of two reactants is called a second-order reaction (molecularity) 2 H H HO C _ _ C Br Br HO H H H H Reaction rates (kinetic) can be expressed mathematically: reaction rate = disappearance of reactants (or appearance of products) For the disappearance of reactants: - rate = k [CH3Br] [OH ] [CH3Br] = CH3Br concentration [OH-] = OH- concentration k= constant (rate constant) L mol•sec For the reaction above, product formation involves a collision between both reactants, thus the rate of the reaction is dependent upon the concentration of both. -

Activation of Alcohols Toward Nucleophilic Substitution: Conversion of Alcohols to Alkyl Halides Amani Atiyalla Abdugadar
University of Northern Colorado Scholarship & Creative Works @ Digital UNC Theses Student Research 12-1-2012 Activation of Alcohols Toward Nucleophilic Substitution: Conversion of Alcohols to Alkyl Halides Amani Atiyalla Abdugadar Follow this and additional works at: http://digscholarship.unco.edu/theses Recommended Citation Abdugadar, Amani Atiyalla, "Activation of Alcohols Toward Nucleophilic Substitution: Conversion of Alcohols to Alkyl Halides" (2012). Theses. Paper 22. This Text is brought to you for free and open access by the Student Research at Scholarship & Creative Works @ Digital UNC. It has been accepted for inclusion in Theses by an authorized administrator of Scholarship & Creative Works @ Digital UNC. For more information, please contact [email protected]. © 2012 Amani Abdugadar ALL RIGHTS RESERVED UNIVERSITY OF NORTHERN COLORADO Greeley, Colorado The Graduate School ACTIVATION OF ALCOHOLS TOWARD NEOCLEOPHILIC SUBSTITUTION: CONVERSION OF ALCOHOLS TO ALKYL HALIDES A Thesis Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Science Amani Abdugadar College of Natural and Health Sciences Department of Chemistry and Biochemistry December, 2012 This Thesis by: Amani Abdugadar Entitled: Activation of Alcohols Toward Neocleophilic Substitution: Conversion of Alcohols to Alkyl Halides has been approved as meeting the requirement for the Master of Science in College of Natural and Health Sciences in Department of Chemistry and Biochemistry Accepted by the Thesis Committee ______________________________________________________ Michael D. Mosher, Ph.D., Research Co-Advisor ______________________________________________________ Richard W. Schwenz, Ph.D., Research Co-Advisor ______________________________________________________ David L. Pringle, Ph.D., Committee Member Accepted by the Graduate School _________________________________________________________ Linda L. Black, Ed.D., LPC Acting Dean of the Graduate School and International Admissions ABSTRACT Abdugadar, Amani. -

Types of Chemical Reactions Rate of a Reaction Factors Affecting Rate of A
B. Sc. II-Sem Rate of formation of a product is positive d[C] Rate of formation of C dt Chemical Kinetics d[D] Rate of formation of D The branch of physical chemistry which deals with the rate at dt which the chemical reactions occur, the mechanism by which the In terms of stoichiometric coefficient rate may be chemical reactions take place and the influence of various factors expressed as dx 1 d[A] 1 d[B] 1 d[C] 1 d[D] such as concentration, temperature, pressure, catalyst etc., on the reaction rates is called the chemical kinetics. dt a dt b dt c dt d dt The rate of reaction is always positive. Types of chemical reactions The rate of chemical reaction decreases as the reaction On the basis of reaction rates, the chemical reactions have proceeds. been classified into the following three types, Unit of conc. Unit of rate of a reaction = =mole L–1 time –1 (1) Very fast or instantaneous reactions: These reactions Unit of time occur at a very fast rate generally these reactions involve ionic In term of gaseous reaction the unit is atm time-1 and species and known as ionic reactions. It is almost impossible to Rate in atm time-1= Rate in mole L1time 1 RT determine the rates of these reactions. Examples (i) AgNO NaCl AgCl NaNO (Precipitation reaction) 3 3 Conc. of product (PPt.) (ii) HCl NaOH NaCl H 2 O (Neutralization reaction) (acid) (base) (Salt) (2) Moderate reaction: These reactions proceed with a measurable rates at normal temperature and it is these reactions (mole/lit.) Conc. -

Model Syllabus Chemistry Revised.Pdf
STATE MODEL SYLLABUS FOR UNDER GRADUATE COURSE IN CHEMISTRY (Bachelor of Science Examination) UNDER CHOICE BASED CREDIT SYSTEM Course structure of UG Chemistry Honours Semester Course Course Name Credits Total marks I AECC-I AECC-I 04 100 C-I Inorganic Chemistry-I 04 75 C-I Practical Inorganic Chemistry-I Lab 02 25 C-II Physical Chemistry-I 04 75 C-II Practical Physical Chemistry-I Lab 02 25 GE-I GE-I 04 75 GE-I Practical GE-I Lab 02 25 22 400 II AECC-II AECC-II 04 100 C-III Organic Chemistry-I 04 75 C-III Practical Organic Chemistry-I Lab 02 25 C-IV Physical Chemistry-II 04 75 C-IV Practical Physical Chemistry-II 02 25 GE-II GE-II 04 75 GE-II Practical GE-II Lab 02 25 22 400 III C-V Inorganic Chemistry-II 04 75 C-V Practical Inorganic Chemistry-II Lab 02 25 C-VI Organic Chemistry-II 04 75 C-VI Practical Organic Chemistry-II Lab 02 25 C-VII Physical Chemistry-III 04 75 C-VII Practical Physical Chemistry-III Lab 02 25 GE-III GE-III 04 75 GE-III Practical GE-III Lab 02 25 SECC-I SECC-I 04 100 28 500 IV C-VIII Inorganic Chemistry-III 04 75 C-VIII Practical Inorganic Chemistry-III Lab 02 25 C-IX Organic Chemistry-III 04 75 C-IX Practical Organic Chemistry-III Lab 02 25 C-X Physical Chemistry-IV 04 75 C-X Practical Physical Chemistry-IV Lab 02 25 GE-IV GE-IV (Theory) 04 75 GE-IV Practical GE-IV (Practical) 02 25 SECC-II SECC-II 04 100 28 500 V C-XI Organic Chemistry-IV 04 75 C-XI Practical Organic Chemistry-IV 02 25 C-XII Physical Chemistry-V 04 75 C-XII Practical Physical Chemistry-V 02 25 DSE-I DSE-I 04 75 DSE-I Practical DSE-I Lab 02 25 DSE-II DSE-II 04 75 DSE-II Practical DSE-II Lab 02 25 24 400 VI C-XIII Inorganic Chemistry- IV 04 75 C-XIII Practical Inorganic Chemistry-IV 02 25 C-XIV Organic Chemistry-V 04 75 C-XIV Practical Organic Chemistry-V 02 25 DSE-III DSE-III 04 75 DSE-III Practical DSE-III Lab 02 25 DSE-IV DSE-IV 04 75 DSE-IV Practical DSE-IV Lab 02 25 OR DSE-IV Dissertation 06 100* 24 400 TOTAL 148 2600 Discipline Specific Elective Papers: (Credit: 06 each) (4 papers to be selected by students of Chemistry Honours): DSE (1-IV) 1. -

Chapter Preview
© No part of this publication should be reproduced, stored in a retrieval system, or transmitted in any form or (Written according to Revised Syllabus of University of Mumbai any means, electronic, mechanical, photocopying, recording and/or otherwise without the prior written with effect from the academic year 2017-18) permission of the publisher. First Edition : 1995 Seventh Revised Edition : 2004 Eighth Revised Edition : 2005 Reprint : 2006, 2007, 2009 COLLEGE Ninth Revised Edition : 2010 Tenth Revised Edition : 2012 Eleventh Revised Edition : 2013 Reprint : 2014 Twelfth Revised Edition : 2015 ORGANIC (As per New Syllabus) Thirteenth Edition : 2016 Fourteenth Revised Edition : 2017 (As per New Syllabus) Fifteenth Edition : 2018 CHEMISTRY Sixteenth Edition : 2019 Published by : Mrs. Meena Pandey for Himalaya Publishing House Pvt. Ltd., S.Y.B.Sc. “Ramdoot”, Dr. Bhalerao Marg, Girgaon, Mumbai - 400 004. Phone: 022-23860170/23863863, Fax: 022-23877178 E-mail: [email protected];Website: www.himpub.com Branch Offices : New Delhi : “Pooja Apartments”, 4-B, Murari Lal Street, Ansari Road, Darya Ganj, New Delhi - 110 002. Phones: 011-23270392, 23278631; Fax: 011-23256286 Nagpur : Kundanlal Chandak Industrial Estate, Ghat Road, R.S. Rao Dr. (Mrs.) Sushil Puniyani Nagpur - 440 018. Phones: 0712-2738731, 3296733; Telefax: 0712-2721215 Vice-Principal & Associate Professor, M.Sc., M.Phil., Ph.D., Bengaluru : Plot No. 91-33, 2nd Main Road, Seshadripuram, Dept. of Chemistry, Rtd. Head, Dept. of Chemistry, Behind Nataraja Theatre, Bengaluru - 560 020. G.N. Khalsa College of Arts, Science & Commerce, K.C. College of Arts, Science and Commerce, Phones: 08041138821, 09379847017, 09379847005 Matunga, Mumbai. Churchgate, Mumbai. Hyderabad : No. 3-4-184, Lingampally, Besides Raghavendra Swamy Matham, Kachiguda, Hyderabad - 500 027. -

Kinetics: Rates and Mechanisms of Chemical Reactions Chapter 13
Chapter 13 Kinetics: Rates and Mechanisms of Chemical Reactions 14.1 Focusing on Reaction Rate 14.2 Expressing the Reaction Rate 14.3 The Rate Law and Its Components 14.4 Integrated Rate Laws: Concentration Changes over Time 14.5 Theories of Chemical Kinetics 14.6 Reaction Mechanisms: The Steps from Reactant to Product 14.7 Catalysis: Speeding Up a Reaction ©2013 McGraw-Hill Ryerson Limited 14-1 Factors That Influence Reaction Rate • Particles must collide in order to react. • The higher the concentration of reactants, the greater the reaction rate. – A higher concentration of reactant particles allows a greater number of collisions. • The physical state of the reactants influences reaction rate. – Substances must mix in order for particles to collide. • The higher the temperature, the greater the reaction rate. – At higher temperatures particles have more energy and therefore collide more often and more effectively. ©2013 McGraw-Hill Ryerson Limited 14-2 Figure 14.3 The effect of surface area on reaction rate. A hot steel nail glows feebly The same mass of steel wool when placed in O2. bursts into flame. ©2013 McGraw-Hill Ryerson Limited 14-3 Figure 14.4 Sufficient collision energy is required for a reaction to occur. ©2013 McGraw-Hill Ryerson Limited 14-4 14.2 Expressing the Reaction Rate Reaction rate is measured in terms of the changes in concentrations of reactants or products per unit time. For the general reaction A → B, we measure the concentration of A at t1 and at t2: change in concentration of A conc A2 - conc A1 Δ(conc Α) Rate = = - = - Δt change in time t2 - t1 Square brackets indicate a concentration in moles per liter. -

Physical Chemistry
Physical Chemistry Lecture 5 Theoretical chemical kinetics Chemical kinetics Understand the nature of reactions Predict reaction outcomes based on Reactants Conditions Requires integration of theory and experimental results Temperature dependence of rate constants k(T ) = Aexp(−Ea / RT) Empirical theory of Arrhenius gives a useful way to parameterize rate constants A ≡ pre-exponential factor Ea ≡ activation energy Often seen in analysis of kinetic data Non-Arrhenius behavior Arrhenius behavior is empirical For some reactions the temperature dependence of the rate constant is not exponential Theory does not easily predict Arrhenius form Amazing that Arrhenius behavior is so often seen Elementary reactions Chemical reactions are often more complex than presented and do not occur as a single step Some reactions do occur in a single step -- elementary reactions Generally involve simple mono- or bimolecular interactions Order in elementary reactions is the stoichiometry number, called molecularity 1 2 H 2 + 2O → H 2O2 v = k[H 2 ] [O] H + Br → HBr v = k[H ]1[Br]1 Simple collision theory of gas- phase kinetics To participate in a bimolecular reaction, v ∝ < Z AB > molecules must * * vmax = < Z AB > = σ AB < vAB > nAnB approach each other 8kT = σ N C C SCT: gas-phase reaction AB 0 πµ A B rate proportional to collision frequency SCT does not generally σ = πd 2 ≡ collision cross − section agree with experimental AB AB rates 8kT k = σ N Points out how to think max AB 0 πµ about theory of chemical reactions “Correcting” simple collision theory