Activation Induced Cytidine Deaminase Mutant
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Activation-Induced Deoxycytidine Deaminase (AID) Co-Transcriptional Scanning at Single-Molecule Resolution
ARTICLE Received 19 Nov 2014 | Accepted 13 Nov 2015 | Published 18 Dec 2015 DOI: 10.1038/ncomms10209 OPEN Activation-induced deoxycytidine deaminase (AID) co-transcriptional scanning at single-molecule resolution Gayan Senavirathne1,*, Jeffrey G. Bertram2,*, Malgorzata Jaszczur2,*, Kathy R. Chaurasiya3,4,*, Phuong Pham2, Chi H. Mak5,6, Myron F. Goodman2,5 & David Rueda1,3,4 Activation-induced deoxycytidine deaminase (AID) generates antibody diversity in B cells by initiating somatic hypermutation (SHM) and class-switch recombination (CSR) during transcription of immunoglobulin variable (IgV) and switch region (IgS) DNA. Using single- molecule FRET, we show that AID binds to transcribed dsDNA and translocates unidirectionally in concert with RNA polymerase (RNAP) on moving transcription bubbles, while increasing the fraction of stalled bubbles. AID scans randomly when constrained in an 8 nt model bubble. When unconstrained on single-stranded (ss) DNA, AID moves in random bidirectional short slides/hops over the entire molecule while remaining bound for B5 min. Our analysis distinguishes dynamic scanning from static ssDNA creasing. That AID alone can track along with RNAP during transcription and scan within stalled transcription bubbles suggests a mechanism by which AID can initiate SHM and CSR when properly regulated, yet when unregulated can access non-Ig genes and cause cancer. 1 Department of Chemistry, Wayne State University, 5101 Cass Avenue, Detroit, Michigan 48202, USA. 2 Department of Biological Sciences, University of Southern California, Los Angeles, California 90089, USA. 3 Department of Medicine, Section of Virology, Imperial College London, Du Cane Road, London W12 0NN, UK. 4 Single Molecule Imaging Group, MRC Clinical Sciences Center, Imperial College London, Du Cane Road, London W12 0NN, UK. -

Difluorodeoxycytidine 5'-Triphosphate: a Mechanism of Self-Potentiation1
ICANCER RESEARCH 52, 533-539, February 1, 1992] Cellular Elimination of 2',2'-Difluorodeoxycytidine 5'-Triphosphate: A Mechanism of Self-Potentiation1 Volker Heinemann,2 Y¡-ZhengXu, Sherri Chubb, Alina Sen, Larry W. Hertel, Gerald B. Grindey, and William Plunkett1 Department of Medical Oncology, The University of Texas M. D. Anderson Cancer Center, Houston, Texas 77030 [V. H., Y. X., S. C., A. S., W. P.], and Lilly Research Laboratories, Indianapolis, Indiana 46285 (L. W. H., G. B. G.] ABSTRACT less, clinical cellular pharmacology studies have demonstrated 2',2'-Difluorodeoxycytidine (dFdC, Gemcitabine) is a deoxycytidine that the dFdCTP:dCTP value reaches potentially inhibitory analogue which, after phosphorylation to the 5'-di- and 5'-triphosphate values during clinical trials (4, 10, 11). (b) dFdCTP is incor porated into DNA by DNA polymerases a and f, inhibiting (dFdCTP), induces inhibition of DNA synthesis and cell death. We examined the values for elimination kinetics of cellular dFdCTP and further elongation (9). (c) Once incorporated, dFdCMP resi found they were dependent on cellular concentration after incubation of dues in the terminal or penultimate positions of the DNA strand CCRF-CEM cells with dFdC and washing into drug-free medium. When inhibit the editing function of DNA polymerase <(9). This may the drug was washed out at low cellular dFdCTP levels (<50 n\\), fix damage caused by the incorporated analogue, (d) dFdCDP dFdCTP elimination was linear (t,: = 3.3 h), but it became biphasic at inhibits ribonucleotide reducÃase,blocking DNA synthesis by intracellular dFdCTP levels >100 JIM. Although the initial elimination decreasing the cellular concentrations of deoxynucleoside tri rate was similar at all concentrations, at higher concentrations the phosphates (12-14). -

B Number Gene Name Mrna Intensity Mrna Present # of Tryptic
list list sample) short list predicted B number Gene name assignment mRNA present mRNA intensity Gene description Protein detected - Membrane protein detected (total list) detected (long list) membrane sample Proteins detected - detected (short list) # of tryptic peptides # of tryptic peptides # of tryptic peptides # of tryptic peptides # of tryptic peptides Functional category detected (membrane Protein detected - total Protein detected - long b0003 thrB 6781 P 9 P 3 3 P 3 0 homoserine kinase Metabolism of small molecules b0004 thrC 15039 P 18 P 10 P 11 P 10 0 threonine synthase Metabolism of small molecules b0008 talB 20561 P 20 P 13 P 16 P 13 0 transaldolase B Metabolism of small molecules b0009 mog 1296 P 7 0 0 0 0 required for the efficient incorporation of molybdate into molybdoproteins Metabolism of small molecules b0014 dnaK 13283 P 32 P 23 P 24 P 23 0 chaperone Hsp70; DNA biosynthesis; autoregulated heat shock proteins Cell processes b0031 dapB 2348 P 16 P 3 3 P 3 0 dihydrodipicolinate reductase Metabolism of small molecules b0032 carA 9312 P 14 P 8 P 8 P 8 0 carbamoyl-phosphate synthetase, glutamine (small) subunit Metabolism of small molecules b0048 folA 1588 P 7 P 1 2 P 1 0 dihydrofolate reductase type I; trimethoprim resistance Metabolism of small molecules peptidyl-prolyl cis-trans isomerase (PPIase), involved in maturation of outer b0053 surA 3825 P 19 P 4 P 5 P 4 P(m) 1 GenProt membrane proteins (1st module) Cell processes b0054 imp 2737 P 42 P 5 0 0 P(m) 5 GenProt organic solvent tolerance Cell processes b0071 leuD 4770 -

Supplementary Information
Supplementary information (a) (b) Figure S1. Resistant (a) and sensitive (b) gene scores plotted against subsystems involved in cell regulation. The small circles represent the individual hits and the large circles represent the mean of each subsystem. Each individual score signifies the mean of 12 trials – three biological and four technical. The p-value was calculated as a two-tailed t-test and significance was determined using the Benjamini-Hochberg procedure; false discovery rate was selected to be 0.1. Plots constructed using Pathway Tools, Omics Dashboard. Figure S2. Connectivity map displaying the predicted functional associations between the silver-resistant gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S3. Connectivity map displaying the predicted functional associations between the silver-sensitive gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S4. Metabolic overview of the pathways in Escherichia coli. The pathways involved in silver-resistance are coloured according to respective normalized score. Each individual score represents the mean of 12 trials – three biological and four technical. Amino acid – upward pointing triangle, carbohydrate – square, proteins – diamond, purines – vertical ellipse, cofactor – downward pointing triangle, tRNA – tee, and other – circle. -

Induction of Deoxycytidine Deaminase Activity in Mammalian Cell Lines By
Proc. Nati. Acad. Sci. USA Vol. 74, No. 4, pp. 1734-1738, April 1977 Microbiology Induction of deoxycytidine deaminase activity in mammalian cell lines by infection with herpes simplex virus type 1 (mouse mutant cell lines/selective system/thymidine kinase/antiviral chemotherapy) TEH-SHENG CHAN Department of Physiology, University of Connecticut Health Center, Farmington, Connecticut 06032 Communicated by E. A. Adelberg, January 26,1977 ABSTRACT Herpes simplex virus type 1 induces deoxycy- activity when it lytically infects dCD- mouse cells. It will also tidine deaminase (cytidine/deoxycytidine aminohydrolase, EC be shown that the induced enzyme is very likely coded by the 3.5.4.5) activity when it lytically infects a number of mammalian is a useful cell lines. The deaminase activity is induced in a mouse cell line viral genome. We have previously shown that dCD that is deficient in this enzyme. The induction of the enzyme selective marker for somatic cell hybridization (14). Thus, this in this mutant cell line does not occur in the presence of acti- enzyme can also be used as a selective marker for isolating nomycin D and the induced enzyme is more thermolabile than dCD+ cells from dCD- cells after infection by UV-inactivated the enzyme.of the wild-type mouse cell line. Furthermore, a new HSV-1 virus. deoxycytidine deaminase species with a characteristic elec- trophoretic mobility that is different from that of the host cell enzyme is found in cell extracts prepard from a human cell line MATERIALS AND METHODS infected with herpesvirus. These results strongly suggest that the virus-induced deoxycytidine deaminase is coded by the viral Cells. -

RNA-Mediated Dimerization of the Human Deoxycytidine Deaminase
Article RNA-Mediated Dimerization of the Human Deoxycytidine Deaminase APOBEC3H Influences Enzyme Activity and Interaction with Nucleic Acids Yuqing Feng 1, Lai Wong 1, Michael Morse 2, Ioulia Rouzina 3, Mark C. Williams 2 and Linda Chelico 1 1 - Department of Biochemistry, Microbiology, and Immunology, University of Saskatchewan, Saskatoon, SK S7N 5E5, Canada 2 - Department of Physics, Northeastern University, Boston, MA 02115, USA 3 - Department of Chemistry and Biochemistry, Ohio State University, Columbus, OH 43210, USA Correspondence to Linda Chelico: [email protected] https://doi.org/10.1016/j.jmb.2018.11.006 Edited by Eric O. Freed Abstract Human APOBEC3H is a single-stranded (ss)DNA deoxycytidine deaminase that inhibits replication of retroelements and HIV-1 in CD4+ T cells. When aberrantly expressed in lung or breast tissue, APOBEC3H can contribute to cancer mutagenesis. These different activities are carried out by different haplotypes of APOBEC3H. Here we studied APOBEC3H haplotype II, which is able to restrict HIV-1 replication and retroelements. We determined how the dimerization mechanism, which is mediated by a double-stranded RNA molecule, influenced interactions with and activity on ssDNA. The data demonstrate that the cellular RNA bound by APOBEC3H does not completely inhibit enzyme activity, in contrast to other APOBEC family members. Despite degradation of the cellular RNA, an approximately 12-nt RNA remains bound to the enzyme, even in the presence of ssDNA. The RNA-mediated dimer is disrupted by mutating W115 on loop 7 or R175 and R176 on helix 6, but this also disrupts protein stability. In contrast, mutation of Y112 and Y113 on loop 7 also destabilizes RNA-mediated dimerization but results in a stable enzyme. -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

Unveiling Human Non-Random Genome Editing Mechanisms Activated in Response to Chronic Environmental Changes
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 19 July 2020 Unveiling human non-random genome editing mechanisms activated in response to chronic environmental changes. I. Where might these mechanisms come from? Loris Zamai Department of Biomolecular Sciences, University of Urbino Carlo Bo; Urbino; National Institute for Nuclear Physics (INFN)-Gran Sasso National Laboratory (LNGS), Assergi, L'Aquila, Italy. Corresponding author: Loris Zamai, Department of Biomolecular Sciences, via Ca’ le Suore 2, University of Urbino Carlo Bo, 61029 Urbino, Italy. Tel. (+39) 0722 304319; fax (+39) 0722 304319; e-mail: [email protected] Running Headline: Non-random genome editing mechanisms Key words: environment, virus, pollutant, evolution, exaptation, stem cells, transposons, LINE-1, Alu, mutagenic enzymes, APOBEC, ADAR, ORF2p, cancer, Eco-Evo-Devo, symbiosis, ecological genomics, environmental stress, genetic recombination, biological plasticity, hypermutation, epigenetics, fractal systems, natural selection. List of Abbreviations single nucleotide polymorphisms, (SNP); copy number variations, (CNV); stress induced mutagenesis, (SIM); ecological evolutionary developmental biology, (Eco-Evo-Devo); somatic hypermutation, (SHM); immunoglobulin, (Ig); activation-induced deaminase (AID); CRISPR, (clustered, regularly interspaced, short, palindromic repeats); Cas, (CRISPR-associated); CRISPR RNAs, (crRNAs); Cascade, (CRISPR-associated complex for antiviral defense); apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like, (APOBEC); -

Nucleoside Analogs: Molecular Mechanisms Signaling Cell Death
Oncogene (2008) 27, 6522–6537 & 2008 Macmillan Publishers Limited All rights reserved 0950-9232/08 $32.00 www.nature.com/onc REVIEW Nucleoside analogs: molecular mechanisms signaling cell death B Ewald, D Sampath and W Plunkett Department of Experimental Therapeutics, The University of Texas MD Anderson Cancer Center, Houston, TX, USA Nucleoside analogs are structurally similar antimeta- clinically active in indolent diseases, in which cells are bolites that have a broad range of action and are clinically not actively replicating. Others, which reverse epigenetic active in both solid tumors and hematological malignan- gene silencing caused by DNA methylation offer an cies. Many of these agents are incorporated into DNAby additional use for these agents during cancer therapy. polymerases during normal DNAsynthesis, an action that This article will address the actions of select nucleoside blocks further extension of the nascent strand and causes analogs that are established as effective cancer therapeu- stalling of replication forks. The molecular mechanisms tics and will briefly mention others that are being that sense stalled replication forks activate cell cycle developed, focusing on mechanisms of action that induce checkpoints and DNArepair processes, which may apoptosis. Subsequently, the current state of knowledge contribute to drug resistance. When replication forks are regarding the cellular and molecular mechanisms that not stabilized by these molecules or when subsequent DNA sense causes for nucleoside analog-induced toxicity is repair processes are overwhelmed, apoptosis is initiated reviewed by proposing signaling models that lead to either by these same DNAdamage sensors or by death signaling, or conversely, spare toxicity. In addition, alternative mechanisms. -

Deoxycytidine Excretion in Vitamin B6 Or Pantothenic Acid Deficient Rats
AN ABSTRACT OF THE THESIS OF CHRISTINE MAY JENSEN for the degree of MASTER OF SCIENCE in Foods and Nutrition presented on May 12,1978 DEOXYCYTIDINE EXCRETION IN VITAMIN B6 OR PANTOTHENIC ACID DEFICIENT RATS Abstract Approved: i ^im W ■■ fm There are two studies on male Wistar rats presented in this thesis. Study I was designed to.quantitate the effect of vitamin B6 or pantothenic acid deficiencies on the urinary excretion of deoxycytidine. Study II was designed to determine: if the decreased deoxycytidine excrertion seen in the vitamin B6 deficient rats of Study I persisted when the control animals were pair fed to the vitamin B6 deficient animals; the effect of a vitamin B6 deficiency on incorporation of deoxycytidine; - and how a vitamin B6 deficiency affects the normal elevation of deoxycytidine excretion in rats which occurs after exposure to X-irradiation. Results from Study I demonstrated that there was a ten fold decrease in deoxycytidine excretion by the third week of the vitamin B6 deficiency and a three fold decrease by the fifth week of the pantothenic acid deficiency. These effects were reversed with repletion of the vitamins. Deoxycytidine excretion returned to normal in the vitamin B6 deficient group by the end of the first week of repletion and in the pantothenic acid deficient group by the end of the second week of repletion. Results from Study II were more difficult to interpret because the control and deficient diets were not comparable. Analysis of the diets demonstrated the control diet to have only one fifth of the vitamin mix which was expected to be in the diet and which was in the deficient diet. -

POLSKIE TOWARZYSTWO BIOCHEMICZNE Postępy Biochemii
POLSKIE TOWARZYSTWO BIOCHEMICZNE Postępy Biochemii http://rcin.org.pl WSKAZÓWKI DLA AUTORÓW Kwartalnik „Postępy Biochemii” publikuje artykuły monograficzne omawiające wąskie tematy, oraz artykuły przeglądowe referujące szersze zagadnienia z biochemii i nauk pokrewnych. Artykuły pierwszego typu winny w sposób syntetyczny omawiać wybrany temat na podstawie możliwie pełnego piśmiennictwa z kilku ostatnich lat, a artykuły drugiego typu na podstawie piśmiennictwa z ostatnich dwu lat. Objętość takich artykułów nie powinna przekraczać 25 stron maszynopisu (nie licząc ilustracji i piśmiennictwa). Kwartalnik publikuje także artykuły typu minireviews, do 10 stron maszynopisu, z dziedziny zainteresowań autora, opracowane na podstawie najnow szego piśmiennictwa, wystarczającego dla zilustrowania problemu. Ponadto kwartalnik publikuje krótkie noty, do 5 stron maszynopisu, informujące o nowych, interesujących osiągnięciach biochemii i nauk pokrewnych, oraz noty przybliżające historię badań w zakresie różnych dziedzin biochemii. Przekazanie artykułu do Redakcji jest równoznaczne z oświadczeniem, że nadesłana praca nie była i nie będzie publikowana w innym czasopiśmie, jeżeli zostanie ogłoszona w „Postępach Biochemii”. Autorzy artykułu odpowiadają za prawidłowość i ścisłość podanych informacji. Autorów obowiązuje korekta autorska. Koszty zmian tekstu w korekcie (poza poprawieniem błędów drukarskich) ponoszą autorzy. Artykuły honoruje się według obowiązujących stawek. Autorzy otrzymują bezpłatnie 25 odbitek swego artykułu; zamówienia na dodatkowe odbitki (płatne) należy zgłosić pisemnie odsyłając pracę po korekcie autorskiej. Redakcja prosi autorów o przestrzeganie następujących wskazówek: Forma maszynopisu: maszynopis pracy i wszelkie załączniki należy nadsyłać w dwu egzem plarzach. Maszynopis powinien być napisany jednostronnie, z podwójną interlinią, z marginesem ok. 4 cm po lewej i ok. 1 cm po prawej stronie; nie może zawierać więcej niż 60 znaków w jednym wierszu nie więcej niż 30 wierszy na stronie zgodnie z Normą Polską. -
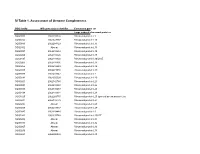
SI Table 1. Assessment of Genome Completeness
SI Table 1. Assessment of Genome Completeness COG family IMG gene object identifier Conserved gene set Large subunit ribosomal proteins COG0081 2062288324 Ribosomal protein L1 COG0244 2062347387 Ribosomal protein L10 COG0080 2062288323 Ribosomal protein L11 COG0102 Absent Ribosomal protein L13 COG0093 2062418832 Ribosomal protein L14 COG0200 2062418826 Ribosomal protein L15 COG0197 2062418838 Ribosomal protein L16/L10E COG0203 2062418836 Ribosomal protein L17 COG0256 2062418829 Ribosomal protein L18 COG0335 2062273558 Ribosomal protein L19 COG0090 2062418842 Ribosomal protein L2 COG0292 2062350539 Ribosomal protein L20 COG0261 2062142780 Ribosomal protein L21 COG0091 2062418840 Ribosomal protein L22 COG0089 2062138283 Ribosomal protein L23 COG0198 2062418834 Ribosomal protein L24 COG1825 2062269715 Ribosomal protein L25 (general stress protein Ctc) COG0211 2062142779 Ribosomal protein L27 COG0227 Absent Ribosomal protein L28 COG0255 2062418837 Ribosomal protein L29 COG0087 2062154483 Ribosomal protein L3 COG1841 2062335748 Ribosomal protein L30/L7E COG0254 Absent Ribosomal protein L31 COG0333 Absent Ribosomal protein L32 COG0267 Absent Ribosomal protein L33 COG0230 Absent Ribosomal protein L34 COG0291 2062350538 Ribosomal protein L35 COG0257 Absent Ribosomal protein L36 COG0088 2062138282 Ribosomal protein L4 COG0094 2062418833 Ribosomal protein L5 COG0097 2062418830 Ribosomal protein L6P/L9E COG0222 2062288326 Ribosomal protein L7/L12 COG0359 2062209880 Ribosomal protein L9 Small subunit ribosomal proteins COG0539 Absent Ribosomal protein