Oligohydramnios Precision Panel Overview Indications Clinical Utility
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A CRISPR-Cas9–Engineered Mouse Model for GPI-Anchor Deficiency Mirrors Human Phenotypes and Exhibits Hippocampal Synaptic Dysfunctions
A CRISPR-Cas9–engineered mouse model for GPI-anchor deficiency mirrors human phenotypes and exhibits hippocampal synaptic dysfunctions Miguel Rodríguez de los Santosa,b,c,d, Marion Rivalane,f, Friederike S. Davidd,g, Alexander Stumpfh, Julika Pitschi,j, Despina Tsortouktzidisi, Laura Moreno Velasquezh, Anne Voigth, Karl Schillingk, Daniele Matteil, Melissa Longe,f, Guido Vogta,c, Alexej Knausd, Björn Fischer-Zirnsaka,c, Lars Wittlerm, Bernd Timmermannn, Peter N. Robinsono,p, Denise Horna, Stefan Mundlosa,c, Uwe Kornaka,c,q, Albert J. Beckeri, Dietmar Schmitzh, York Wintere,f, and Peter M. Krawitzd,1 aInstitute for Medical Genetics and Human Genetics, Charité–Universitätsmedizin Berlin, 13353 Berlin, Germany; bBerlin-Brandenburg School for Regenerative Therapies, Charité-Universitätsmedizin Berlin, 13353 Berlin, Germany; cResearch Group Development and Disease, Max Planck Institute for Molecular Genetics, 14195 Berlin, Germany; dInstitute for Genomic Statistics and Bioinformatics, University of Bonn, 53127 Bonn, Germany; eAnimal Outcome Core Facility of the NeuroCure Center, Charité–Universitätsmedizin Berlin, 10117 Berlin, Germany; fInstitute of Cognitive Neurobiology, Humboldt University, 10117 Berlin, Germany; gInstitute of Human Genetics, Faculty of Medicine, University Hospital Bonn, 53127 Bonn, Germany; hNeuroscience Research Center, Charité–Universitätsmedizin Berlin, 10117 Berlin, Germany; iSection for Translational Epilepsy Research, Department of Neuropathology, University Hospital Bonn, 53127 Bonn, Germany; jDepartment of Epileptology, -

Cldn19 Clic2 Clmp Cln3
NewbornDx™ Advanced Sequencing Evaluation When time to diagnosis matters, the NewbornDx™ Advanced Sequencing Evaluation from Athena Diagnostics delivers rapid, 5- to 7-day results on a targeted 1,722-genes. A2ML1 ALAD ATM CAV1 CLDN19 CTNS DOCK7 ETFB FOXC2 GLUL HOXC13 JAK3 AAAS ALAS2 ATP1A2 CBL CLIC2 CTRC DOCK8 ETFDH FOXE1 GLYCTK HOXD13 JUP AARS2 ALDH18A1 ATP1A3 CBS CLMP CTSA DOK7 ETHE1 FOXE3 GM2A HPD KANK1 AASS ALDH1A2 ATP2B3 CC2D2A CLN3 CTSD DOLK EVC FOXF1 GMPPA HPGD K ANSL1 ABAT ALDH3A2 ATP5A1 CCDC103 CLN5 CTSK DPAGT1 EVC2 FOXG1 GMPPB HPRT1 KAT6B ABCA12 ALDH4A1 ATP5E CCDC114 CLN6 CUBN DPM1 EXOC4 FOXH1 GNA11 HPSE2 KCNA2 ABCA3 ALDH5A1 ATP6AP2 CCDC151 CLN8 CUL4B DPM2 EXOSC3 FOXI1 GNAI3 HRAS KCNB1 ABCA4 ALDH7A1 ATP6V0A2 CCDC22 CLP1 CUL7 DPM3 EXPH5 FOXL2 GNAO1 HSD17B10 KCND2 ABCB11 ALDOA ATP6V1B1 CCDC39 CLPB CXCR4 DPP6 EYA1 FOXP1 GNAS HSD17B4 KCNE1 ABCB4 ALDOB ATP7A CCDC40 CLPP CYB5R3 DPYD EZH2 FOXP2 GNE HSD3B2 KCNE2 ABCB6 ALG1 ATP8A2 CCDC65 CNNM2 CYC1 DPYS F10 FOXP3 GNMT HSD3B7 KCNH2 ABCB7 ALG11 ATP8B1 CCDC78 CNTN1 CYP11B1 DRC1 F11 FOXRED1 GNPAT HSPD1 KCNH5 ABCC2 ALG12 ATPAF2 CCDC8 CNTNAP1 CYP11B2 DSC2 F13A1 FRAS1 GNPTAB HSPG2 KCNJ10 ABCC8 ALG13 ATR CCDC88C CNTNAP2 CYP17A1 DSG1 F13B FREM1 GNPTG HUWE1 KCNJ11 ABCC9 ALG14 ATRX CCND2 COA5 CYP1B1 DSP F2 FREM2 GNS HYDIN KCNJ13 ABCD3 ALG2 AUH CCNO COG1 CYP24A1 DST F5 FRMD7 GORAB HYLS1 KCNJ2 ABCD4 ALG3 B3GALNT2 CCS COG4 CYP26C1 DSTYK F7 FTCD GP1BA IBA57 KCNJ5 ABHD5 ALG6 B3GAT3 CCT5 COG5 CYP27A1 DTNA F8 FTO GP1BB ICK KCNJ8 ACAD8 ALG8 B3GLCT CD151 COG6 CYP27B1 DUOX2 F9 FUCA1 GP6 ICOS KCNK3 ACAD9 ALG9 -

Blueprint Genetics Comprehensive Growth Disorders / Skeletal
Comprehensive Growth Disorders / Skeletal Dysplasias and Disorders Panel Test code: MA4301 Is a 374 gene panel that includes assessment of non-coding variants. This panel covers the majority of the genes listed in the Nosology 2015 (PMID: 26394607) and all genes in our Malformation category that cause growth retardation, short stature or skeletal dysplasia and is therefore a powerful diagnostic tool. It is ideal for patients suspected to have a syndromic or an isolated growth disorder or a skeletal dysplasia. About Comprehensive Growth Disorders / Skeletal Dysplasias and Disorders This panel covers a broad spectrum of diseases associated with growth retardation, short stature or skeletal dysplasia. Many of these conditions have overlapping features which can make clinical diagnosis a challenge. Genetic diagnostics is therefore the most efficient way to subtype the diseases and enable individualized treatment and management decisions. Moreover, detection of causative mutations establishes the mode of inheritance in the family which is essential for informed genetic counseling. For additional information regarding the conditions tested on this panel, please refer to the National Organization for Rare Disorders and / or GeneReviews. Availability 4 weeks Gene Set Description Genes in the Comprehensive Growth Disorders / Skeletal Dysplasias and Disorders Panel and their clinical significance Gene Associated phenotypes Inheritance ClinVar HGMD ACAN# Spondyloepimetaphyseal dysplasia, aggrecan type, AD/AR 20 56 Spondyloepiphyseal dysplasia, Kimberley -

Prenatalscreen® Standard Technical Report
About PrenatalScreen® Prenatal Test PrenatalScreen® Prenatal Test is a genetic test that analyses fetal DNA, obtained from CVS or amniotic fluid following an invasive prenatal diagnosis, to screen for monogenic disorders in the fetus. Using the latest technologies, including Next Generation Sequencing (NGS), PrenatalScreen® Prenatal Test screen 744 genes for mutations causing over 1.000 severe genetic disorders in the fetus. PrenatalScreen® Prenatal Test allows for a comprehensive care and enables patients to make more informed reproductive decisions. Offering PrenatalScreen® Prenatal Test to a patient during pregnancy allows her to gain more knowledge about the potential to pass along a condition to the fetus. Aim of the test PrenatalScreen® Prenatal Test analyses DNA extracted from fetal cells in the amniotic fluid, collected through amniocentesis, or in the chorionic villi through villocentesis (CVS). The aim of this diagnositc test is to assess severe genetic diseases in the fetus, including the most common diseases in the European population. Genes listed in Table 1 were selected according to the incidence in the population of the disease caused by mutations in such genes, the severity of the clinical phenotype at birth and the importance of the related pathogenetic picture, in accordance with the indications of the American College of Medical Genetics (ACMG)(Grody et al., Genet Med 2013:15:482–483). PrenatalScreen®: Indication for testing PrenatalScreen® Prenatal Test is intended for patients who meet any of the following criteria: • Personal/familial anamnesis of hereditary genetic diseases; • For expectant mothers wishing to reduce the risk of a genetic diseases in the fetus; • For natural or in vitro fertilization (IVF)-derived pregnancies: • For couples using heterologus IVF procedures (egg/sperm donors). -

The Genomic Basis of Evolved Virus Resistance Is Dependent on Environmental Resources
bioRxiv preprint doi: https://doi.org/10.1101/666404; this version posted June 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 The genomic basis of evolved virus resistance is dependent on environmental 2 resources 3 4 Katherine Roberts1¶, Sean Meaden 1 ¶, Stephen Sharpe, Suzanne Kay, Toby Doyle, Drew 5 Wilson, Lewis J. Bartlett4, Steve Paterson3, Mike Boots1,2* 6 7 1. Biosciences, University of Exeter, Penryn Campus, UK. TR10 9FE 8 2. Integrative Biology, University of California, Berkeley, USA. CA 94720 9 3. Institute of Integrative Biology, University of Liverpool, UK. L69 7ZB 10 4. Department of Biology, Emory University, Atlanta, GA, 30322, USA 11 ¶ These authors contributed equally to this work 12 * Corresponding author 13 14 15 16 17 18 19 20 21 22 23 24 25 1 bioRxiv preprint doi: https://doi.org/10.1101/666404; this version posted June 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 26 Abstract 27 28 Parasites impose strong selection on their hosts, but the level of resistance evolved may be 29 constrained by the availability of resources. However, studies identifying the genomic basis 30 of such resource mediated selection are rare, particularly in non-model organisms. Here, we 31 investigated the role of nutrition in the evolution of resistance to a DNA virus (PiGV), and 32 associated trade-offs, in a lepidopteran pest species (Plodia interpunctella). -

Blueprint Genetics Comprehensive Skeletal Dysplasias and Disorders
Comprehensive Skeletal Dysplasias and Disorders Panel Test code: MA3301 Is a 251 gene panel that includes assessment of non-coding variants. Is ideal for patients with a clinical suspicion of disorders involving the skeletal system. About Comprehensive Skeletal Dysplasias and Disorders This panel covers a broad spectrum of skeletal disorders including common and rare skeletal dysplasias (eg. achondroplasia, COL2A1 related dysplasias, diastrophic dysplasia, various types of spondylo-metaphyseal dysplasias), various ciliopathies with skeletal involvement (eg. short rib-polydactylies, asphyxiating thoracic dysplasia dysplasias and Ellis-van Creveld syndrome), various subtypes of osteogenesis imperfecta, campomelic dysplasia, slender bone dysplasias, dysplasias with multiple joint dislocations, chondrodysplasia punctata group of disorders, neonatal osteosclerotic dysplasias, osteopetrosis and related disorders, abnormal mineralization group of disorders (eg hypopohosphatasia), osteolysis group of disorders, disorders with disorganized development of skeletal components, overgrowth syndromes with skeletal involvement, craniosynostosis syndromes, dysostoses with predominant craniofacial involvement, dysostoses with predominant vertebral involvement, patellar dysostoses, brachydactylies, some disorders with limb hypoplasia-reduction defects, ectrodactyly with and without other manifestations, polydactyly-syndactyly-triphalangism group of disorders, and disorders with defects in joint formation and synostoses. Availability 4 weeks Gene Set Description -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Sema4 Cardiac Information Sheet
CARDIAC NEXT-GENERATION SEQUENCING PANELS Mail: One Gustave L. Levy Place, Box 1497 1 CLIA #: 33D2097541 Specimens: 1428 Madison Ave, Atran Bldg, Rm 2-25 T: 800-298-6470 New York, NY 10029 F: 212-241-0139 www.sema4.com 0 3 8 Mail: One Gustave L. Levy Place, Box 1497 2 CLIA #: 33D2097541 Specimens: 1428 Madison Ave, Atran Bldg, Rm 2-25 T: 800-298-6470 New York, NY 10029 F: 212-241-0139 www.sema4.com Mail: One Gustave L. Levy Place, Box 1497 3 CLIA #: 33D2097541 Specimens: 1428 Madison Ave, Atran Bldg, Rm 2-25 T: 800-298-6470 New York, NY 10029 F: 212-241-0139 www.sema4.com Mail: One Gustave L. Levy Place, Box 1497 4 CLIA #: 33D2097541 Specimens: 1428 Madison Ave, Atran Bldg, Rm 2-25 T: 800-298-6470 New York, NY 10029 F: 212-241-0139 www.sema4.com TABLE OF CONTENTS GENETIC TESTING FOR INHERITED CARDIOVASCULAR CONDITIONS 6 GENETICS AND INDICATIONS 6 TESTING METHODS, SENSITIVITY, AND LIMITATIONS 7 TURNAROUND TIME 9 SPECIMEN AND SHIPPING REQUIREMENTS 9 CUSTOMER SERVICES AND GENETIC COUNSELING 10 THE COMPREHENSIVE CARDIOMYOPATHY PANEL 11 DILATED CARDIOMYOPATHY (DCM) SUBPANEL 23 ARRHYTHMOGENIC RIGHT VENTRICULAR CARDIOMYOPATHY (ARVC) SUBPANEL 28 HYPERTROPHIC CARDIOMYOPATHY (HCM) SUBPANEL 30 LEFT VENTRICULAR NON-COMPACTION CARDIOMYOPATHY (LVNC) SUBPANEL 33 THE COMPREHENSIVE ARRHYTHMIAS PANEL 35 BRUGADA SYNDROME (BRS) SUBPANEL 40 CATECHOLAMINERGIC POLYMORPHIC VENTRICULAR TACHYCARDIA (CPVT) SUBPANEL 42 LONG / SHORT QT SYNDROME (LQTS / SQTS) SUBPANEL 44 AORTOPATHIES PANEL 46 CONGENITAL HEART DISEASE (CHD) PANEL 49 FAMILIAL HYPERCHOLESTEROLEMIA (FH) PANEL 52 PULMONARY HYPERTENSION (PAH) PANEL 54 METABOLIC CARDIOMYOPATHY PANEL 55 NOONAN SPECTRUM DISORDERS PANEL 57 HEREDITARY HEMORRHAGIC TELANGIECTASIA PANEL 59 REFERENCES 60 DISCLAIMER 69 Mail: One Gustave L. -

Transcriptome Profiling and Molecular Pathway Analysis of Genes in Association with Salinity Adaptation in Nile Tilapia Oreochromis Niloticus
RESEARCH ARTICLE Transcriptome Profiling and Molecular Pathway Analysis of Genes in Association with Salinity Adaptation in Nile Tilapia Oreochromis niloticus Zhixin Xu1, Lei Gan1, Tongyu Li1, Chang Xu1, Ke Chen1, Xiaodan Wang1, Jian G. Qin2, Liqiao Chen1*, Erchao Li1* 1 Laboratory of Aquaculture Nutrition and Environmental Health, School of Life Sciences, East China Normal University, 500 Dongchuan Rd., Shanghai 200241, China, 2 School of Biological Sciences, Flinders University, Adelaide, SA 5001, Australia * [email protected] (EL); [email protected] (LC) Abstract Nile tilapia Oreochromis niloticus is a freshwater fish but can tolerate a wide range of salini- OPEN ACCESS ties. The mechanism of salinity adaptation at the molecular level was studied using RNA- Citation: Xu Z, Gan L, Li T, Xu C, Chen K, Wang X, Seq to explore the molecular pathways in fish exposed to 0, 8, or 16 (practical salinity unit, et al. (2015) Transcriptome Profiling and Molecular psu). Based on the change of gene expressions, the differential genes unions from freshwa- Pathway Analysis of Genes in Association with Salinity Adaptation in Nile Tilapia Oreochromis ter to saline water were classified into three categories. In the constant change category (1), niloticus. PLoS ONE 10(8): e0136506. doi:10.1371/ steroid biosynthesis, steroid hormone biosynthesis, fat digestion and absorption, comple- journal.pone.0136506 ment and coagulation cascades were significantly affected by salinity indicating the pivotal Editor: Marie-Joelle Virolle, University Paris South, roles of sterol-related pathways in response to salinity stress. In the change-then-stable cat- FRANCE egory (2), ribosomes, oxidative phosphorylation, signaling pathways for peroxisome prolif- Received: June 4, 2015 erator activated receptors, and fat digestion and absorption changed significantly with Accepted: August 4, 2015 increasing salinity, showing sensitivity to salinity variation in the environment and a responding threshold to salinity change. -

The Phenotype/Genotype Relation and the Current QT
110 Heart 1997;78:1 10-116 REVIEW The phenotype/genotype relation and the current status of genetic screening in hypertrophic cardiomyopathy, Marfan syndrome, and the long QT syndrome J Burn, J Camm, M J Davies, L Peltonen, P J Schwartz, H Watkins Hypertrophic cardiomyopathy, Marfan syn- encode amino acids, but within the gene there drome, and the long QT syndrome are all are also non-functional sequences (introns). autosomal disorders inherited in a dominant The simplest single gene abnormality is a manner; affected individuals are heterozygous, point mutation in which one base pair is that is they have one normal and one mutant replaced by another. Because of the redun- copy of the gene but not all the gene carriers dancy in the genetic code, point mutations are symptomatic. This failure to express fully may be silent polymorphisms that do not the expected phenotype is traditionally known change the amino acid or they may be mis- as incomplete penetrance. sense mutations that change a single amino acid. Missense mutations can be regarded as analogous to simple spelling errors in one let- Vocabulary and technology ter of a word. For example "the fish is on the In the human genome (consisting of 22 pairs dish" becomes "the fish is on the fish". of autosomes and the X and Y chromosomes) Because each gene exists in a maternal and a there are between 50 000 and 100 000 indi- paternal copy the ultimate product of the gene vidual genes coding for a wide range of prod- is a combination of normal (wild type) and ucts including enzymes and structural abnormal (mutant type). -
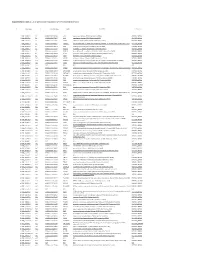
Supplemetary Table 2. List of Genes Down-Regulated in LPAR6 Knocked Down Cells
Supplemetary Table 2. List of genes down-regulated in LPAR6 knocked down cells g# initial alias c# converted alias name description namespace 1 NM_002317.5 1.1 ENSG00000113083 LOX lysyl oxidase [Source:HGNC Symbol;Acc:6664] REFSEQ_MRNA 2 NM_006183.4 2.1 ENSG00000133636 NTS neurotensin [Source:HGNC Symbol;Acc:8038] REFSEQ_MRNA 3 NM_005213.3 3.1 ENSG00000121552 CSTA cystatin A (stefin A) [Source:HGNC Symbol;Acc:2481] REFSEQ_MRNA 4 NM_007231.3 4.1 ENSG00000087916 SLC6A14 solute carrier family 6 (amino acid transporter), member 14 [Source:HGNC Symbol;Acc:11047] REFSEQ_MRNA 5 NM_001873.2 5.1 ENSG00000109472 CPE carboxypeptidase E [Source:HGNC Symbol;Acc:2303] REFSEQ_MRNA 6 NM_019856.1 6.1 ENSG00000101605 MYOM1 myomesin 1, 185kDa [Source:HGNC Symbol;Acc:7613] REFSEQ_MRNA 7 NM_032590.4 7.1 ENSG00000089094 KDM2B lysine (K)-specific demethylase 2B [Source:HGNC Symbol;Acc:13610] REFSEQ_MRNA 8 NM_001901.2 8.1 ENSG00000118523 CTGF connective tissue growth factor [Source:HGNC Symbol;Acc:2500] REFSEQ_MRNA 9 NM_007183.2 9.1 ENSG00000184363 PKP3 plakophilin 3 [Source:HGNC Symbol;Acc:9025] REFSEQ_MRNA 10 NM_182965.2 10.1 ENSG00000176170 SPHK1 sphingosine kinase 1 [Source:HGNC Symbol;Acc:11240] REFSEQ_MRNA 11 NM_152423.4 11.1 ENSG00000157502 MUM1L1 melanoma associated antigen (mutated) 1-like 1 [Source:HGNC Symbol;Acc:26583] REFSEQ_MRNA 12 NM_002923.3 12.1 ENSG00000116741 RGS2 regulator of G-protein signaling 2, 24kDa [Source:HGNC Symbol;Acc:9998] REFSEQ_MRNA 13 NR_003038.2 13.1 N/A N/A N/A N/A 14 NM_080862.1 14.1 ENSG00000175093 SPSB4 splA/ryanodine receptor -

CENTOGENE's Severe and Early Onset Disorder Gene List
CENTOGENE’s severe and early onset disorder gene list USED IN PRENATAL WES ANALYSIS AND IDENTIFICATION OF “PATHOGENIC” AND “LIKELY PATHOGENIC” CENTOMD® VARIANTS IN NGS PRODUCTS The following gene list shows all genes assessed in prenatal WES tests or analysed for P/LP CentoMD® variants in NGS products after April 1st, 2020. For searching a single gene coverage, just use the search on www.centoportal.com AAAS, AARS1, AARS2, ABAT, ABCA12, ABCA3, ABCB11, ABCB4, ABCB7, ABCC6, ABCC8, ABCC9, ABCD1, ABCD4, ABHD12, ABHD5, ACACA, ACAD9, ACADM, ACADS, ACADVL, ACAN, ACAT1, ACE, ACO2, ACOX1, ACP5, ACSL4, ACTA1, ACTA2, ACTB, ACTG1, ACTL6B, ACTN2, ACVR2B, ACVRL1, ACY1, ADA, ADAM17, ADAMTS2, ADAMTSL2, ADAR, ADARB1, ADAT3, ADCY5, ADGRG1, ADGRG6, ADGRV1, ADK, ADNP, ADPRHL2, ADSL, AFF2, AFG3L2, AGA, AGK, AGL, AGPAT2, AGPS, AGRN, AGT, AGTPBP1, AGTR1, AGXT, AHCY, AHDC1, AHI1, AIFM1, AIMP1, AIPL1, AIRE, AK2, AKR1D1, AKT1, AKT2, AKT3, ALAD, ALDH18A1, ALDH1A3, ALDH3A2, ALDH4A1, ALDH5A1, ALDH6A1, ALDH7A1, ALDOA, ALDOB, ALG1, ALG11, ALG12, ALG13, ALG14, ALG2, ALG3, ALG6, ALG8, ALG9, ALMS1, ALOX12B, ALPL, ALS2, ALX3, ALX4, AMACR, AMER1, AMN, AMPD1, AMPD2, AMT, ANK2, ANK3, ANKH, ANKRD11, ANKS6, ANO10, ANO5, ANOS1, ANTXR1, ANTXR2, AP1B1, AP1S1, AP1S2, AP3B1, AP3B2, AP4B1, AP4E1, AP4M1, AP4S1, APC2, APTX, AR, ARCN1, ARFGEF2, ARG1, ARHGAP31, ARHGDIA, ARHGEF9, ARID1A, ARID1B, ARID2, ARL13B, ARL3, ARL6, ARL6IP1, ARMC4, ARMC9, ARSA, ARSB, ARSL, ARV1, ARX, ASAH1, ASCC1, ASH1L, ASL, ASNS, ASPA, ASPH, ASPM, ASS1, ASXL1, ASXL2, ASXL3, ATAD3A, ATCAY, ATIC, ATL1, ATM, ATOH7,