A CRISPR-Cas9–Engineered Mouse Model for GPI-Anchor Deficiency Mirrors Human Phenotypes and Exhibits Hippocampal Synaptic Dysfunctions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Cldn19 Clic2 Clmp Cln3
NewbornDx™ Advanced Sequencing Evaluation When time to diagnosis matters, the NewbornDx™ Advanced Sequencing Evaluation from Athena Diagnostics delivers rapid, 5- to 7-day results on a targeted 1,722-genes. A2ML1 ALAD ATM CAV1 CLDN19 CTNS DOCK7 ETFB FOXC2 GLUL HOXC13 JAK3 AAAS ALAS2 ATP1A2 CBL CLIC2 CTRC DOCK8 ETFDH FOXE1 GLYCTK HOXD13 JUP AARS2 ALDH18A1 ATP1A3 CBS CLMP CTSA DOK7 ETHE1 FOXE3 GM2A HPD KANK1 AASS ALDH1A2 ATP2B3 CC2D2A CLN3 CTSD DOLK EVC FOXF1 GMPPA HPGD K ANSL1 ABAT ALDH3A2 ATP5A1 CCDC103 CLN5 CTSK DPAGT1 EVC2 FOXG1 GMPPB HPRT1 KAT6B ABCA12 ALDH4A1 ATP5E CCDC114 CLN6 CUBN DPM1 EXOC4 FOXH1 GNA11 HPSE2 KCNA2 ABCA3 ALDH5A1 ATP6AP2 CCDC151 CLN8 CUL4B DPM2 EXOSC3 FOXI1 GNAI3 HRAS KCNB1 ABCA4 ALDH7A1 ATP6V0A2 CCDC22 CLP1 CUL7 DPM3 EXPH5 FOXL2 GNAO1 HSD17B10 KCND2 ABCB11 ALDOA ATP6V1B1 CCDC39 CLPB CXCR4 DPP6 EYA1 FOXP1 GNAS HSD17B4 KCNE1 ABCB4 ALDOB ATP7A CCDC40 CLPP CYB5R3 DPYD EZH2 FOXP2 GNE HSD3B2 KCNE2 ABCB6 ALG1 ATP8A2 CCDC65 CNNM2 CYC1 DPYS F10 FOXP3 GNMT HSD3B7 KCNH2 ABCB7 ALG11 ATP8B1 CCDC78 CNTN1 CYP11B1 DRC1 F11 FOXRED1 GNPAT HSPD1 KCNH5 ABCC2 ALG12 ATPAF2 CCDC8 CNTNAP1 CYP11B2 DSC2 F13A1 FRAS1 GNPTAB HSPG2 KCNJ10 ABCC8 ALG13 ATR CCDC88C CNTNAP2 CYP17A1 DSG1 F13B FREM1 GNPTG HUWE1 KCNJ11 ABCC9 ALG14 ATRX CCND2 COA5 CYP1B1 DSP F2 FREM2 GNS HYDIN KCNJ13 ABCD3 ALG2 AUH CCNO COG1 CYP24A1 DST F5 FRMD7 GORAB HYLS1 KCNJ2 ABCD4 ALG3 B3GALNT2 CCS COG4 CYP26C1 DSTYK F7 FTCD GP1BA IBA57 KCNJ5 ABHD5 ALG6 B3GAT3 CCT5 COG5 CYP27A1 DTNA F8 FTO GP1BB ICK KCNJ8 ACAD8 ALG8 B3GLCT CD151 COG6 CYP27B1 DUOX2 F9 FUCA1 GP6 ICOS KCNK3 ACAD9 ALG9 -

High Mutation Frequency of the PIGA Gene in T Cells Results In
High Mutation Frequency of the PIGA Gene in T Cells Results in Reconstitution of GPI A nchor−/CD52− T Cells That Can Give Early Immune Protection after This information is current as Alemtuzumab-Based T Cell−Depleted of October 1, 2021. Allogeneic Stem Cell Transplantation Floris C. Loeff, J. H. Frederik Falkenburg, Lois Hageman, Wesley Huisman, Sabrina A. J. Veld, H. M. Esther van Egmond, Marian van de Meent, Peter A. von dem Borne, Hendrik Veelken, Constantijn J. M. Halkes and Inge Jedema Downloaded from J Immunol published online 2 February 2018 http://www.jimmunol.org/content/early/2018/02/02/jimmun ol.1701018 http://www.jimmunol.org/ Supplementary http://www.jimmunol.org/content/suppl/2018/02/02/jimmunol.170101 Material 8.DCSupplemental Why The JI? Submit online. by guest on October 1, 2021 • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2018 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published February 2, 2018, doi:10.4049/jimmunol.1701018 The Journal of Immunology High Mutation Frequency of the PIGA Gene in T Cells Results in Reconstitution of GPI Anchor2/CD522 T Cells That Can Give Early Immune Protection after Alemtuzumab- Based T Cell–Depleted Allogeneic Stem Cell Transplantation Floris C. -

Comparing Spatial Expression Dynamics of Bovine
Comparing spatial expression dynamics of bovine blastocyst under three different procedures : in-vivo, in-vitro derived, Title and somatic cell nuclear transfer embryos Nagatomo, Hiroaki; Akizawa, Hiroki; Sada, Ayari; Kishi, Yasunori; Yamanaka, Ken-ichi; Takuma, Tetsuya; Sasaki, Author(s) Keisuke; Yamauchi, Nobuhiko; Yanagawa, Yojiro; Nagano, Masashi; Kono, Tomohiro; Takahashi, Masashi; Kawahara, Manabu Citation Japanese Journal of Veterinary Research, 63(4), 159-171 Issue Date 2015-11 DOI 10.14943/jjvr.63.4.159 Doc URL http://hdl.handle.net/2115/60304 Type bulletin (article) Additional Information There are other files related to this item in HUSCAP. Check the above URL. File Information JJVR63-4 p.159-171 Suppl. Table 3.pdf (Supplemental Table 3) Instructions for use Hokkaido University Collection of Scholarly and Academic Papers : HUSCAP Supplemental table 3. Genes that were differentially expressed in the ICM relative to the TE in in SCNT blastocyst (SCNT list). Gene Symbol Probe Set ID Regulation Fold change ([ICM] / [TE]) Gene Title EEF1A1 AFFX-Bt-ef1a-3_at UP 1.2092365 eukaryotic translation elongation factor 1 alpha 1 IGFBP3 Bt.422.1.S2_at UP 2.5323892 insulin-like growth factor binding protein 3 IGFBP3 Bt.422.1.S1_at UP 3.7850845 insulin-like growth factor binding protein 3 SULT1A1 Bt.3537.1.S1_at UP 2.7092714 sulfotransferase family, cytosolic, 1A, phenol-preferring, member 1 SPP1 Bt.2632.1.S1_at UP 5.6928325 secreted phosphoprotein 1 SCARB1 Bt.4520.1.S1_at UP 3.106944 scavenger receptor class B, member 1 TSPO Bt.3988.1.S1_at -

The Genomic Basis of Evolved Virus Resistance Is Dependent on Environmental Resources
bioRxiv preprint doi: https://doi.org/10.1101/666404; this version posted June 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 The genomic basis of evolved virus resistance is dependent on environmental 2 resources 3 4 Katherine Roberts1¶, Sean Meaden 1 ¶, Stephen Sharpe, Suzanne Kay, Toby Doyle, Drew 5 Wilson, Lewis J. Bartlett4, Steve Paterson3, Mike Boots1,2* 6 7 1. Biosciences, University of Exeter, Penryn Campus, UK. TR10 9FE 8 2. Integrative Biology, University of California, Berkeley, USA. CA 94720 9 3. Institute of Integrative Biology, University of Liverpool, UK. L69 7ZB 10 4. Department of Biology, Emory University, Atlanta, GA, 30322, USA 11 ¶ These authors contributed equally to this work 12 * Corresponding author 13 14 15 16 17 18 19 20 21 22 23 24 25 1 bioRxiv preprint doi: https://doi.org/10.1101/666404; this version posted June 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 26 Abstract 27 28 Parasites impose strong selection on their hosts, but the level of resistance evolved may be 29 constrained by the availability of resources. However, studies identifying the genomic basis 30 of such resource mediated selection are rare, particularly in non-model organisms. Here, we 31 investigated the role of nutrition in the evolution of resistance to a DNA virus (PiGV), and 32 associated trade-offs, in a lepidopteran pest species (Plodia interpunctella). -

Complement and Inflammasome Overactivation Mediates Paroxysmal Nocturnal Hemoglobinuria with Autoinflammation
Complement and inflammasome overactivation mediates paroxysmal nocturnal hemoglobinuria with autoinflammation Britta Höchsmann, … , Peter M. Krawitz, Taroh Kinoshita J Clin Invest. 2019. https://doi.org/10.1172/JCI123501. Research Article Hematology Inflammation Graphical abstract Find the latest version: https://jci.me/123501/pdf The Journal of Clinical Investigation RESEARCH ARTICLE Complement and inflammasome overactivation mediates paroxysmal nocturnal hemoglobinuria with autoinflammation Britta Höchsmann,1,2 Yoshiko Murakami,3,4 Makiko Osato,3,5 Alexej Knaus,6 Michi Kawamoto,7 Norimitsu Inoue,8 Tetsuya Hirata,3 Shogo Murata,3,9 Markus Anliker,1 Thomas Eggermann,10 Marten Jäger,11 Ricarda Floettmann,11 Alexander Höllein,12 Sho Murase,7 Yasutaka Ueda,5 Jun-ichi Nishimura,5 Yuzuru Kanakura,5 Nobuo Kohara,7 Hubert Schrezenmeier,1 Peter M. Krawitz,6 and Taroh Kinoshita3,4 1Institute of Transfusion Medicine, University of Ulm, Ulm, Germany. 2Institute of Clinical Transfusion Medicine and Immunogenetics, German Red Cross Blood Transfusion Service and University Hospital Ulm, Ulm, Germany. 3Research Institute for Microbial Diseases and 4WPI Immunology Frontier Research Center, Osaka University, Osaka, Japan. 5Department of Hematology and Oncology, Graduate School of Medicine, Osaka University, Osaka, Japan. 6Institute for Genomic Statistics and Bioinformatics, Rheinische Friedrich-Wilhelms-Universität Bonn, Bonn, Germany. 7Department of Neurology, Kobe City Medical Center General Hospital, Kobe, Japan. 8Department of Tumor Immunology, Osaka -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Transcriptome Profiling and Molecular Pathway Analysis of Genes in Association with Salinity Adaptation in Nile Tilapia Oreochromis Niloticus
RESEARCH ARTICLE Transcriptome Profiling and Molecular Pathway Analysis of Genes in Association with Salinity Adaptation in Nile Tilapia Oreochromis niloticus Zhixin Xu1, Lei Gan1, Tongyu Li1, Chang Xu1, Ke Chen1, Xiaodan Wang1, Jian G. Qin2, Liqiao Chen1*, Erchao Li1* 1 Laboratory of Aquaculture Nutrition and Environmental Health, School of Life Sciences, East China Normal University, 500 Dongchuan Rd., Shanghai 200241, China, 2 School of Biological Sciences, Flinders University, Adelaide, SA 5001, Australia * [email protected] (EL); [email protected] (LC) Abstract Nile tilapia Oreochromis niloticus is a freshwater fish but can tolerate a wide range of salini- OPEN ACCESS ties. The mechanism of salinity adaptation at the molecular level was studied using RNA- Citation: Xu Z, Gan L, Li T, Xu C, Chen K, Wang X, Seq to explore the molecular pathways in fish exposed to 0, 8, or 16 (practical salinity unit, et al. (2015) Transcriptome Profiling and Molecular psu). Based on the change of gene expressions, the differential genes unions from freshwa- Pathway Analysis of Genes in Association with Salinity Adaptation in Nile Tilapia Oreochromis ter to saline water were classified into three categories. In the constant change category (1), niloticus. PLoS ONE 10(8): e0136506. doi:10.1371/ steroid biosynthesis, steroid hormone biosynthesis, fat digestion and absorption, comple- journal.pone.0136506 ment and coagulation cascades were significantly affected by salinity indicating the pivotal Editor: Marie-Joelle Virolle, University Paris South, roles of sterol-related pathways in response to salinity stress. In the change-then-stable cat- FRANCE egory (2), ribosomes, oxidative phosphorylation, signaling pathways for peroxisome prolif- Received: June 4, 2015 erator activated receptors, and fat digestion and absorption changed significantly with Accepted: August 4, 2015 increasing salinity, showing sensitivity to salinity variation in the environment and a responding threshold to salinity change. -
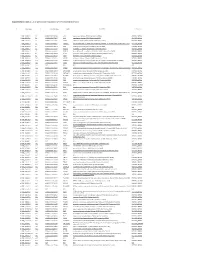
Supplemetary Table 2. List of Genes Down-Regulated in LPAR6 Knocked Down Cells
Supplemetary Table 2. List of genes down-regulated in LPAR6 knocked down cells g# initial alias c# converted alias name description namespace 1 NM_002317.5 1.1 ENSG00000113083 LOX lysyl oxidase [Source:HGNC Symbol;Acc:6664] REFSEQ_MRNA 2 NM_006183.4 2.1 ENSG00000133636 NTS neurotensin [Source:HGNC Symbol;Acc:8038] REFSEQ_MRNA 3 NM_005213.3 3.1 ENSG00000121552 CSTA cystatin A (stefin A) [Source:HGNC Symbol;Acc:2481] REFSEQ_MRNA 4 NM_007231.3 4.1 ENSG00000087916 SLC6A14 solute carrier family 6 (amino acid transporter), member 14 [Source:HGNC Symbol;Acc:11047] REFSEQ_MRNA 5 NM_001873.2 5.1 ENSG00000109472 CPE carboxypeptidase E [Source:HGNC Symbol;Acc:2303] REFSEQ_MRNA 6 NM_019856.1 6.1 ENSG00000101605 MYOM1 myomesin 1, 185kDa [Source:HGNC Symbol;Acc:7613] REFSEQ_MRNA 7 NM_032590.4 7.1 ENSG00000089094 KDM2B lysine (K)-specific demethylase 2B [Source:HGNC Symbol;Acc:13610] REFSEQ_MRNA 8 NM_001901.2 8.1 ENSG00000118523 CTGF connective tissue growth factor [Source:HGNC Symbol;Acc:2500] REFSEQ_MRNA 9 NM_007183.2 9.1 ENSG00000184363 PKP3 plakophilin 3 [Source:HGNC Symbol;Acc:9025] REFSEQ_MRNA 10 NM_182965.2 10.1 ENSG00000176170 SPHK1 sphingosine kinase 1 [Source:HGNC Symbol;Acc:11240] REFSEQ_MRNA 11 NM_152423.4 11.1 ENSG00000157502 MUM1L1 melanoma associated antigen (mutated) 1-like 1 [Source:HGNC Symbol;Acc:26583] REFSEQ_MRNA 12 NM_002923.3 12.1 ENSG00000116741 RGS2 regulator of G-protein signaling 2, 24kDa [Source:HGNC Symbol;Acc:9998] REFSEQ_MRNA 13 NR_003038.2 13.1 N/A N/A N/A N/A 14 NM_080862.1 14.1 ENSG00000175093 SPSB4 splA/ryanodine receptor -

CENTOGENE's Severe and Early Onset Disorder Gene List
CENTOGENE’s severe and early onset disorder gene list USED IN PRENATAL WES ANALYSIS AND IDENTIFICATION OF “PATHOGENIC” AND “LIKELY PATHOGENIC” CENTOMD® VARIANTS IN NGS PRODUCTS The following gene list shows all genes assessed in prenatal WES tests or analysed for P/LP CentoMD® variants in NGS products after April 1st, 2020. For searching a single gene coverage, just use the search on www.centoportal.com AAAS, AARS1, AARS2, ABAT, ABCA12, ABCA3, ABCB11, ABCB4, ABCB7, ABCC6, ABCC8, ABCC9, ABCD1, ABCD4, ABHD12, ABHD5, ACACA, ACAD9, ACADM, ACADS, ACADVL, ACAN, ACAT1, ACE, ACO2, ACOX1, ACP5, ACSL4, ACTA1, ACTA2, ACTB, ACTG1, ACTL6B, ACTN2, ACVR2B, ACVRL1, ACY1, ADA, ADAM17, ADAMTS2, ADAMTSL2, ADAR, ADARB1, ADAT3, ADCY5, ADGRG1, ADGRG6, ADGRV1, ADK, ADNP, ADPRHL2, ADSL, AFF2, AFG3L2, AGA, AGK, AGL, AGPAT2, AGPS, AGRN, AGT, AGTPBP1, AGTR1, AGXT, AHCY, AHDC1, AHI1, AIFM1, AIMP1, AIPL1, AIRE, AK2, AKR1D1, AKT1, AKT2, AKT3, ALAD, ALDH18A1, ALDH1A3, ALDH3A2, ALDH4A1, ALDH5A1, ALDH6A1, ALDH7A1, ALDOA, ALDOB, ALG1, ALG11, ALG12, ALG13, ALG14, ALG2, ALG3, ALG6, ALG8, ALG9, ALMS1, ALOX12B, ALPL, ALS2, ALX3, ALX4, AMACR, AMER1, AMN, AMPD1, AMPD2, AMT, ANK2, ANK3, ANKH, ANKRD11, ANKS6, ANO10, ANO5, ANOS1, ANTXR1, ANTXR2, AP1B1, AP1S1, AP1S2, AP3B1, AP3B2, AP4B1, AP4E1, AP4M1, AP4S1, APC2, APTX, AR, ARCN1, ARFGEF2, ARG1, ARHGAP31, ARHGDIA, ARHGEF9, ARID1A, ARID1B, ARID2, ARL13B, ARL3, ARL6, ARL6IP1, ARMC4, ARMC9, ARSA, ARSB, ARSL, ARV1, ARX, ASAH1, ASCC1, ASH1L, ASL, ASNS, ASPA, ASPH, ASPM, ASS1, ASXL1, ASXL2, ASXL3, ATAD3A, ATCAY, ATIC, ATL1, ATM, ATOH7, -

NGS Catalog: a Database of Next Generation Sequencing Studies in Humans
HUMAN MUTATION Database in Brief 33: E2341-E2355 (2012) Online DATABASE IN BRIEF HUMAN MUTATION OFFICIAL JOURNAL NGS Catalog: A Database of Next Generation Sequencing Studies in Humans www.hgvs.org Junfeng Xia1,†, Qingguo Wang1,†, Peilin Jia1, Bing Wang1, William Pao2,3, and Zhongming Zhao1,3,4,* 1Department of Biomedical Informatics, Vanderbilt University School of Medicine, Nashville, TN, USA,2Department of Medicine/Division of Hematology-Oncology, Vanderbilt University School of Medicine, Nashville, TN, USA,3Vanderbilt-Ingram Cancer Center, Vanderbilt University Medical Center, Nashville, TN, USA,4Department of Psychiatry, Vanderbilt University School of Medicine, Nashville, TN, USA †These authors contributed equally to this work. *Correspondence to Zhongming Zhao, Ph.D., Department of Biomedical Informatics, Vanderbilt University School of Medicine, 2525 West End Avenue, Suite 600, Nashville, TN, 37203, USA; Phone: +1-615-343-9158; FAX: +1-615-936-8545; E-mail: [email protected]. Contract grant sponsor: This study was partially supported by the Stand Up To Cancer-American Association for Cancer Research Innovative Research Grant (SU2C-AACR-IRG0109) and the VICC Cancer Center Core grant P30CA68485 from National Institutes of Health. Communicated by Johan T. den Dunnen ABSTRACT: Next generation sequencing (NGS) technologies have been rapidly applied in biomedical and biological research since its advent only a few years ago, and they are expected to advance at an unprecedented pace in the following years. To provide the research community with a comprehensive NGS resource, we have developed the database Next Generation Sequencing Catalog (NGS Catalog, http://bioinfo.mc.vanderbilt.edu/NGS/index.html), a continually updated database that collects, curates and manages available human NGS data obtained from published literature. -

Downloaded from Here
bioRxiv preprint doi: https://doi.org/10.1101/017566; this version posted November 19, 2015. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 1 Testing for ancient selection using cross-population allele 2 frequency differentiation 1;∗ 3 Fernando Racimo 4 1 Department of Integrative Biology, University of California, Berkeley, CA, USA 5 ∗ E-mail: [email protected] 6 1 Abstract 7 A powerful way to detect selection in a population is by modeling local allele frequency changes in a 8 particular region of the genome under scenarios of selection and neutrality, and finding which model is 9 most compatible with the data. Chen et al. [2010] developed a composite likelihood method called XP- 10 CLR that uses an outgroup population to detect departures from neutrality which could be compatible 11 with hard or soft sweeps, at linked sites near a beneficial allele. However, this method is most sensitive 12 to recent selection and may miss selective events that happened a long time ago. To overcome this, 13 we developed an extension of XP-CLR that jointly models the behavior of a selected allele in a three- 14 population tree. Our method - called 3P-CLR - outperforms XP-CLR when testing for selection that 15 occurred before two populations split from each other, and can distinguish between those events and 16 events that occurred specifically in each of the populations after the split.