Transcriptome Profiling and Molecular Pathway Analysis of Genes in Association with Salinity Adaptation in Nile Tilapia Oreochromis Niloticus
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Enzymatic Encoding Methods for Efficient Synthesis Of
(19) TZZ__T (11) EP 1 957 644 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C12N 15/10 (2006.01) C12Q 1/68 (2006.01) 01.12.2010 Bulletin 2010/48 C40B 40/06 (2006.01) C40B 50/06 (2006.01) (21) Application number: 06818144.5 (86) International application number: PCT/DK2006/000685 (22) Date of filing: 01.12.2006 (87) International publication number: WO 2007/062664 (07.06.2007 Gazette 2007/23) (54) ENZYMATIC ENCODING METHODS FOR EFFICIENT SYNTHESIS OF LARGE LIBRARIES ENZYMVERMITTELNDE KODIERUNGSMETHODEN FÜR EINE EFFIZIENTE SYNTHESE VON GROSSEN BIBLIOTHEKEN PROCEDES DE CODAGE ENZYMATIQUE DESTINES A LA SYNTHESE EFFICACE DE BIBLIOTHEQUES IMPORTANTES (84) Designated Contracting States: • GOLDBECH, Anne AT BE BG CH CY CZ DE DK EE ES FI FR GB GR DK-2200 Copenhagen N (DK) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • DE LEON, Daen SK TR DK-2300 Copenhagen S (DK) Designated Extension States: • KALDOR, Ditte Kievsmose AL BA HR MK RS DK-2880 Bagsvaerd (DK) • SLØK, Frank Abilgaard (30) Priority: 01.12.2005 DK 200501704 DK-3450 Allerød (DK) 02.12.2005 US 741490 P • HUSEMOEN, Birgitte Nystrup DK-2500 Valby (DK) (43) Date of publication of application: • DOLBERG, Johannes 20.08.2008 Bulletin 2008/34 DK-1674 Copenhagen V (DK) • JENSEN, Kim Birkebæk (73) Proprietor: Nuevolution A/S DK-2610 Rødovre (DK) 2100 Copenhagen 0 (DK) • PETERSEN, Lene DK-2100 Copenhagen Ø (DK) (72) Inventors: • NØRREGAARD-MADSEN, Mads • FRANCH, Thomas DK-3460 Birkerød (DK) DK-3070 Snekkersten (DK) • GODSKESEN, -

A CRISPR-Cas9–Engineered Mouse Model for GPI-Anchor Deficiency Mirrors Human Phenotypes and Exhibits Hippocampal Synaptic Dysfunctions
A CRISPR-Cas9–engineered mouse model for GPI-anchor deficiency mirrors human phenotypes and exhibits hippocampal synaptic dysfunctions Miguel Rodríguez de los Santosa,b,c,d, Marion Rivalane,f, Friederike S. Davidd,g, Alexander Stumpfh, Julika Pitschi,j, Despina Tsortouktzidisi, Laura Moreno Velasquezh, Anne Voigth, Karl Schillingk, Daniele Matteil, Melissa Longe,f, Guido Vogta,c, Alexej Knausd, Björn Fischer-Zirnsaka,c, Lars Wittlerm, Bernd Timmermannn, Peter N. Robinsono,p, Denise Horna, Stefan Mundlosa,c, Uwe Kornaka,c,q, Albert J. Beckeri, Dietmar Schmitzh, York Wintere,f, and Peter M. Krawitzd,1 aInstitute for Medical Genetics and Human Genetics, Charité–Universitätsmedizin Berlin, 13353 Berlin, Germany; bBerlin-Brandenburg School for Regenerative Therapies, Charité-Universitätsmedizin Berlin, 13353 Berlin, Germany; cResearch Group Development and Disease, Max Planck Institute for Molecular Genetics, 14195 Berlin, Germany; dInstitute for Genomic Statistics and Bioinformatics, University of Bonn, 53127 Bonn, Germany; eAnimal Outcome Core Facility of the NeuroCure Center, Charité–Universitätsmedizin Berlin, 10117 Berlin, Germany; fInstitute of Cognitive Neurobiology, Humboldt University, 10117 Berlin, Germany; gInstitute of Human Genetics, Faculty of Medicine, University Hospital Bonn, 53127 Bonn, Germany; hNeuroscience Research Center, Charité–Universitätsmedizin Berlin, 10117 Berlin, Germany; iSection for Translational Epilepsy Research, Department of Neuropathology, University Hospital Bonn, 53127 Bonn, Germany; jDepartment of Epileptology, -

Investigation of Plasma Metabolomics and Neurotransmitter Dysfunction In
RSC Advances View Article Online PAPER View Journal | View Issue Investigation of plasma metabolomics and neurotransmitter dysfunction in the process of Cite this: RSC Adv.,2019,9,18308 Alzheimer's disease rat induced by amyloid beta 25- 35 Mengying Wei,ab Yuanyuan Liu,a Zifeng Pi, b Kexin Yue,a Shizhe Li,c Mingxin Hu,a Zhiqiang Liu,b Fengrui Song b and Zhongying Liu*a Alzheimer's disease (AD) has become one of the major diseases endangering the health of the elderly. Clarifying the features of each AD animal model is valuable for understanding the onset and progression of diseases and developing potential treatments in the pharmaceutical industry. In this study, we aimed to clarify plasma metabolomics and neurotransmitter dysfunction in the process of AD model rat induced by amyloid beta 25-35 (Ab 25-35). Firstly, Morris Water Maze (MWM) test was used to investigate cognitive impairment in AD rat after 2, 4 and 8 weeks of modelling. Based on this, the effects on levels Creative Commons Attribution-NonCommercial 3.0 Unported Licence. of AD-related enzymes and eight neurotransmitters were analyzed. And plasma metabolomics analysis based on ultra high-performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry (UHPLC-Q-TOF-MS) was used to research the metabolic disturbances in the process of AD rat. The results shown the injury on the spatial learning ability of AD rats was gradually aggravated within 4 weeks, reached the maximum at 4 weeks and then was stable until 8 weeks. During 8 weeks of modeling, the levels of enzymes including b-secretase, g-secretase, glycogen synthase kinase-3b (GSK- 3b), acetyl cholinesterase (AchE) and nitric oxide synthase (NOS) were significant increased in the plasma of AD rats. -

Cldn19 Clic2 Clmp Cln3
NewbornDx™ Advanced Sequencing Evaluation When time to diagnosis matters, the NewbornDx™ Advanced Sequencing Evaluation from Athena Diagnostics delivers rapid, 5- to 7-day results on a targeted 1,722-genes. A2ML1 ALAD ATM CAV1 CLDN19 CTNS DOCK7 ETFB FOXC2 GLUL HOXC13 JAK3 AAAS ALAS2 ATP1A2 CBL CLIC2 CTRC DOCK8 ETFDH FOXE1 GLYCTK HOXD13 JUP AARS2 ALDH18A1 ATP1A3 CBS CLMP CTSA DOK7 ETHE1 FOXE3 GM2A HPD KANK1 AASS ALDH1A2 ATP2B3 CC2D2A CLN3 CTSD DOLK EVC FOXF1 GMPPA HPGD K ANSL1 ABAT ALDH3A2 ATP5A1 CCDC103 CLN5 CTSK DPAGT1 EVC2 FOXG1 GMPPB HPRT1 KAT6B ABCA12 ALDH4A1 ATP5E CCDC114 CLN6 CUBN DPM1 EXOC4 FOXH1 GNA11 HPSE2 KCNA2 ABCA3 ALDH5A1 ATP6AP2 CCDC151 CLN8 CUL4B DPM2 EXOSC3 FOXI1 GNAI3 HRAS KCNB1 ABCA4 ALDH7A1 ATP6V0A2 CCDC22 CLP1 CUL7 DPM3 EXPH5 FOXL2 GNAO1 HSD17B10 KCND2 ABCB11 ALDOA ATP6V1B1 CCDC39 CLPB CXCR4 DPP6 EYA1 FOXP1 GNAS HSD17B4 KCNE1 ABCB4 ALDOB ATP7A CCDC40 CLPP CYB5R3 DPYD EZH2 FOXP2 GNE HSD3B2 KCNE2 ABCB6 ALG1 ATP8A2 CCDC65 CNNM2 CYC1 DPYS F10 FOXP3 GNMT HSD3B7 KCNH2 ABCB7 ALG11 ATP8B1 CCDC78 CNTN1 CYP11B1 DRC1 F11 FOXRED1 GNPAT HSPD1 KCNH5 ABCC2 ALG12 ATPAF2 CCDC8 CNTNAP1 CYP11B2 DSC2 F13A1 FRAS1 GNPTAB HSPG2 KCNJ10 ABCC8 ALG13 ATR CCDC88C CNTNAP2 CYP17A1 DSG1 F13B FREM1 GNPTG HUWE1 KCNJ11 ABCC9 ALG14 ATRX CCND2 COA5 CYP1B1 DSP F2 FREM2 GNS HYDIN KCNJ13 ABCD3 ALG2 AUH CCNO COG1 CYP24A1 DST F5 FRMD7 GORAB HYLS1 KCNJ2 ABCD4 ALG3 B3GALNT2 CCS COG4 CYP26C1 DSTYK F7 FTCD GP1BA IBA57 KCNJ5 ABHD5 ALG6 B3GAT3 CCT5 COG5 CYP27A1 DTNA F8 FTO GP1BB ICK KCNJ8 ACAD8 ALG8 B3GLCT CD151 COG6 CYP27B1 DUOX2 F9 FUCA1 GP6 ICOS KCNK3 ACAD9 ALG9 -

(10) Patent No.: US 8119385 B2
US008119385B2 (12) United States Patent (10) Patent No.: US 8,119,385 B2 Mathur et al. (45) Date of Patent: Feb. 21, 2012 (54) NUCLEICACIDS AND PROTEINS AND (52) U.S. Cl. ........................................ 435/212:530/350 METHODS FOR MAKING AND USING THEMI (58) Field of Classification Search ........................ None (75) Inventors: Eric J. Mathur, San Diego, CA (US); See application file for complete search history. Cathy Chang, San Diego, CA (US) (56) References Cited (73) Assignee: BP Corporation North America Inc., Houston, TX (US) OTHER PUBLICATIONS c Mount, Bioinformatics, Cold Spring Harbor Press, Cold Spring Har (*) Notice: Subject to any disclaimer, the term of this bor New York, 2001, pp. 382-393.* patent is extended or adjusted under 35 Spencer et al., “Whole-Genome Sequence Variation among Multiple U.S.C. 154(b) by 689 days. Isolates of Pseudomonas aeruginosa” J. Bacteriol. (2003) 185: 1316 1325. (21) Appl. No.: 11/817,403 Database Sequence GenBank Accession No. BZ569932 Dec. 17. 1-1. 2002. (22) PCT Fled: Mar. 3, 2006 Omiecinski et al., “Epoxide Hydrolase-Polymorphism and role in (86). PCT No.: PCT/US2OO6/OOT642 toxicology” Toxicol. Lett. (2000) 1.12: 365-370. S371 (c)(1), * cited by examiner (2), (4) Date: May 7, 2008 Primary Examiner — James Martinell (87) PCT Pub. No.: WO2006/096527 (74) Attorney, Agent, or Firm — Kalim S. Fuzail PCT Pub. Date: Sep. 14, 2006 (57) ABSTRACT (65) Prior Publication Data The invention provides polypeptides, including enzymes, structural proteins and binding proteins, polynucleotides US 201O/OO11456A1 Jan. 14, 2010 encoding these polypeptides, and methods of making and using these polynucleotides and polypeptides. -

The Genomic Basis of Evolved Virus Resistance Is Dependent on Environmental Resources
bioRxiv preprint doi: https://doi.org/10.1101/666404; this version posted June 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 The genomic basis of evolved virus resistance is dependent on environmental 2 resources 3 4 Katherine Roberts1¶, Sean Meaden 1 ¶, Stephen Sharpe, Suzanne Kay, Toby Doyle, Drew 5 Wilson, Lewis J. Bartlett4, Steve Paterson3, Mike Boots1,2* 6 7 1. Biosciences, University of Exeter, Penryn Campus, UK. TR10 9FE 8 2. Integrative Biology, University of California, Berkeley, USA. CA 94720 9 3. Institute of Integrative Biology, University of Liverpool, UK. L69 7ZB 10 4. Department of Biology, Emory University, Atlanta, GA, 30322, USA 11 ¶ These authors contributed equally to this work 12 * Corresponding author 13 14 15 16 17 18 19 20 21 22 23 24 25 1 bioRxiv preprint doi: https://doi.org/10.1101/666404; this version posted June 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 26 Abstract 27 28 Parasites impose strong selection on their hosts, but the level of resistance evolved may be 29 constrained by the availability of resources. However, studies identifying the genomic basis 30 of such resource mediated selection are rare, particularly in non-model organisms. Here, we 31 investigated the role of nutrition in the evolution of resistance to a DNA virus (PiGV), and 32 associated trade-offs, in a lepidopteran pest species (Plodia interpunctella). -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

(12) United States Patent (10) Patent No.: US 7,670,797 B2 Vacanti Et Al
USOO7670797B2 (12) United States Patent (10) Patent No.: US 7,670,797 B2 Vacanti et al. (45) Date of Patent: Mar. 2, 2010 (54) METHOD OF DETERMINING TOXICITY 6,139,574. A 10/2000 Vacanti et al. WITH THREE DIMIENSIONAL STRUCTURES 6,197.575 B1* 3/2001 Griffith et al. ............ 435/288.4 (75) Inventors: Joseph P. Vacanti, Boston, MA (US); 6,287,558 B1 9, 2001 Lanza ettal. a Robert Rubin, Brookline, MA (US); 6,372.482 B1 4/2002 Mitrani et al. Wing Cheung Swampscott MA (US); 6,376, 169 B1 4/2002 Adams et al. Jeffrey T. Borenstein, Newton, MA 6,455,311 B1 9/2002 Vacanti (US) 6.479,064 B1 1 1/2002 Atala 6,525,242 B1 2/2003 Wu et al. (73) Assignees: The General Hospital Corporation, 6542.858 B1 4/2003 Grass et al. 5. R. R. The classert 6,547.994 B1 4/2003 Monkhouse et al. re ry, 2C, 6,573,063 B2 6/2003 Hochman 6,607.910 B1* 8/2003 Dimitrijevich et al. ... 435/297.1 (*) Notice: Subject to any disclaimer, the term of this 6,645,715 B1 1 1/2003 Griffith et al. patent is extended or adjusted under 35 6,647,358 B2 11/2003 Grass et al. U.S.C. 154(b) by 70 days. 2001/0041964 A1 11/2001 Grass et al. (21) Appl. No.: 11/183,115 2002fOO 10550 A1 1/2002 Grass et al. ppl. No.: 9 2002fOO 13662 A1 1/2002 Grass et al. (22)22) Filed: L. 15,9 2005 2002.0035459 A1 3/2002 Grass et al. -
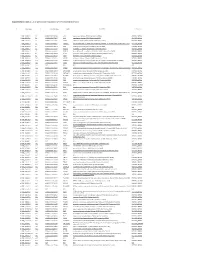
Supplemetary Table 2. List of Genes Down-Regulated in LPAR6 Knocked Down Cells
Supplemetary Table 2. List of genes down-regulated in LPAR6 knocked down cells g# initial alias c# converted alias name description namespace 1 NM_002317.5 1.1 ENSG00000113083 LOX lysyl oxidase [Source:HGNC Symbol;Acc:6664] REFSEQ_MRNA 2 NM_006183.4 2.1 ENSG00000133636 NTS neurotensin [Source:HGNC Symbol;Acc:8038] REFSEQ_MRNA 3 NM_005213.3 3.1 ENSG00000121552 CSTA cystatin A (stefin A) [Source:HGNC Symbol;Acc:2481] REFSEQ_MRNA 4 NM_007231.3 4.1 ENSG00000087916 SLC6A14 solute carrier family 6 (amino acid transporter), member 14 [Source:HGNC Symbol;Acc:11047] REFSEQ_MRNA 5 NM_001873.2 5.1 ENSG00000109472 CPE carboxypeptidase E [Source:HGNC Symbol;Acc:2303] REFSEQ_MRNA 6 NM_019856.1 6.1 ENSG00000101605 MYOM1 myomesin 1, 185kDa [Source:HGNC Symbol;Acc:7613] REFSEQ_MRNA 7 NM_032590.4 7.1 ENSG00000089094 KDM2B lysine (K)-specific demethylase 2B [Source:HGNC Symbol;Acc:13610] REFSEQ_MRNA 8 NM_001901.2 8.1 ENSG00000118523 CTGF connective tissue growth factor [Source:HGNC Symbol;Acc:2500] REFSEQ_MRNA 9 NM_007183.2 9.1 ENSG00000184363 PKP3 plakophilin 3 [Source:HGNC Symbol;Acc:9025] REFSEQ_MRNA 10 NM_182965.2 10.1 ENSG00000176170 SPHK1 sphingosine kinase 1 [Source:HGNC Symbol;Acc:11240] REFSEQ_MRNA 11 NM_152423.4 11.1 ENSG00000157502 MUM1L1 melanoma associated antigen (mutated) 1-like 1 [Source:HGNC Symbol;Acc:26583] REFSEQ_MRNA 12 NM_002923.3 12.1 ENSG00000116741 RGS2 regulator of G-protein signaling 2, 24kDa [Source:HGNC Symbol;Acc:9998] REFSEQ_MRNA 13 NR_003038.2 13.1 N/A N/A N/A N/A 14 NM_080862.1 14.1 ENSG00000175093 SPSB4 splA/ryanodine receptor -

CENTOGENE's Severe and Early Onset Disorder Gene List
CENTOGENE’s severe and early onset disorder gene list USED IN PRENATAL WES ANALYSIS AND IDENTIFICATION OF “PATHOGENIC” AND “LIKELY PATHOGENIC” CENTOMD® VARIANTS IN NGS PRODUCTS The following gene list shows all genes assessed in prenatal WES tests or analysed for P/LP CentoMD® variants in NGS products after April 1st, 2020. For searching a single gene coverage, just use the search on www.centoportal.com AAAS, AARS1, AARS2, ABAT, ABCA12, ABCA3, ABCB11, ABCB4, ABCB7, ABCC6, ABCC8, ABCC9, ABCD1, ABCD4, ABHD12, ABHD5, ACACA, ACAD9, ACADM, ACADS, ACADVL, ACAN, ACAT1, ACE, ACO2, ACOX1, ACP5, ACSL4, ACTA1, ACTA2, ACTB, ACTG1, ACTL6B, ACTN2, ACVR2B, ACVRL1, ACY1, ADA, ADAM17, ADAMTS2, ADAMTSL2, ADAR, ADARB1, ADAT3, ADCY5, ADGRG1, ADGRG6, ADGRV1, ADK, ADNP, ADPRHL2, ADSL, AFF2, AFG3L2, AGA, AGK, AGL, AGPAT2, AGPS, AGRN, AGT, AGTPBP1, AGTR1, AGXT, AHCY, AHDC1, AHI1, AIFM1, AIMP1, AIPL1, AIRE, AK2, AKR1D1, AKT1, AKT2, AKT3, ALAD, ALDH18A1, ALDH1A3, ALDH3A2, ALDH4A1, ALDH5A1, ALDH6A1, ALDH7A1, ALDOA, ALDOB, ALG1, ALG11, ALG12, ALG13, ALG14, ALG2, ALG3, ALG6, ALG8, ALG9, ALMS1, ALOX12B, ALPL, ALS2, ALX3, ALX4, AMACR, AMER1, AMN, AMPD1, AMPD2, AMT, ANK2, ANK3, ANKH, ANKRD11, ANKS6, ANO10, ANO5, ANOS1, ANTXR1, ANTXR2, AP1B1, AP1S1, AP1S2, AP3B1, AP3B2, AP4B1, AP4E1, AP4M1, AP4S1, APC2, APTX, AR, ARCN1, ARFGEF2, ARG1, ARHGAP31, ARHGDIA, ARHGEF9, ARID1A, ARID1B, ARID2, ARL13B, ARL3, ARL6, ARL6IP1, ARMC4, ARMC9, ARSA, ARSB, ARSL, ARV1, ARX, ASAH1, ASCC1, ASH1L, ASL, ASNS, ASPA, ASPH, ASPM, ASS1, ASXL1, ASXL2, ASXL3, ATAD3A, ATCAY, ATIC, ATL1, ATM, ATOH7, -

(12) Patent Application Publication (10) Pub. No.: US 2001/0034023 A1 Stanton, JR
US 2001.0034O23A1. (19) United States (12) Patent Application Publication (10) Pub. No.: US 2001/0034023 A1 Stanton, JR. et al. (43) Pub. Date: Oct. 25, 2001 (54) GENE SEQUENCE VARIATIONS WITH Related U.S. Application Data UTILITY IN DETERMINING THE (63) Continuation-in-part of application No. 09/710,467, TREATMENT OF DISEASE, INGENES filed on Nov. 8, 2000, which is a continuation-in-part RELATING TO DRUG PROCESSING of application No. 09/696,482, filed on Oct. 24, 2000. Non-provisional of provisional application No. 60/131,334, filed on Apr. 26, 1999. Non-provisional (76) Inventors: Vincent P. Stanton JR., Belmont, MA of provisional application No. 60/139,440, filed on (US); Martin Zillmann, Shrewsbury, Jun. 15, 1999. MA (US) (30) Foreign Application Priority Data Correspondence Address: Jan. 20, 2000 (WO)........................... PCT/USOO/O1392 EDWARD O. KRUESSER BROBECK PHLEGER & HARRISON Publication Classification 12390 EL CAMINO REAL (51) Int. Cl. ............................. C12O 1/68; G06F 19/00 SAN DIEGO, CA 92130 (US) (52) U.S. Cl. ................................................... 435/6; 702/20 (57) ABSTRACT (21) Appl. No.: 09/733,000 Methods for identifying and utilizing variances in genes relating to efficacy and Safety of medical therapy and other aspects of medical therapy are described, including methods (22) Filed: Dec. 7, 2000 for Selecting an effective treatment. US 2001/0034023 A1 Oct. 25, 2001 GENE SEQUENCE WARIATIONS WITH UTILITY 0005 For some drugs, over 90% of the measurable IN DETERMINING THE TREATMENT OF interSubject variation in Selected pharmacokinetic param DISEASE, INGENES RELATING TO DRUG eters has been shown to be heritable. For a limited number PROCESSING of drugs, DNA sequence variances have been identified in Specific genes that are involved in drug action or metabo RELATED APPLICATIONS lism, and these variances have been shown to account for the 0001. -

Downloaded from Here
bioRxiv preprint doi: https://doi.org/10.1101/017566; this version posted November 19, 2015. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 1 Testing for ancient selection using cross-population allele 2 frequency differentiation 1;∗ 3 Fernando Racimo 4 1 Department of Integrative Biology, University of California, Berkeley, CA, USA 5 ∗ E-mail: [email protected] 6 1 Abstract 7 A powerful way to detect selection in a population is by modeling local allele frequency changes in a 8 particular region of the genome under scenarios of selection and neutrality, and finding which model is 9 most compatible with the data. Chen et al. [2010] developed a composite likelihood method called XP- 10 CLR that uses an outgroup population to detect departures from neutrality which could be compatible 11 with hard or soft sweeps, at linked sites near a beneficial allele. However, this method is most sensitive 12 to recent selection and may miss selective events that happened a long time ago. To overcome this, 13 we developed an extension of XP-CLR that jointly models the behavior of a selected allele in a three- 14 population tree. Our method - called 3P-CLR - outperforms XP-CLR when testing for selection that 15 occurred before two populations split from each other, and can distinguish between those events and 16 events that occurred specifically in each of the populations after the split.