Periodic Table of the Elements
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Absolute and Relative Facial Selectivities in Organocatalytic Asymmetric Chlorocyclization Reactions
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Absolute and relative facial selectivities in organocatalytic asymmetric chlorocyclization Cite this: Chem. Sci.,2018,9, 2898 reactions† Nastaran Salehi Marzijarani,a Roozbeh Yousefi,a Arvind Jaganathan,b Kumar Dilip Ashtekar,a James E. Jackson *a and Babak Borhan *a Though (DHQD)2PHAL-catalyzed chlorocyclizations of 1,1-disubstituted olefins show useful (and in some cases, reversible) asymmetric induction, stereochemically complete descriptions of these alkene additions have remained largely unknown. Herein, based on a combination of NMR, derivative, isotope labeling, and computational studies, we present detailed stereochemical analyses of chlorocyclizations of nucleophile-tethered 1,1-disubstituted styryl systems. The selectivities of the two asymmetric bond- forming processes, namely electrophilic chlorine attack and nucleophilic ring closure, are thus mapped out independently. Under the established optimal conditions, four related chlorocyclizations were + Creative Commons Attribution 3.0 Unported Licence. subjected to this analysis. All showed a strong preference for Cl delivery from the same face of the Received 12th October 2017 alkene. However, depending on reaction conditions and substrate identity (carboxylic acid, amide or Accepted 24th December 2017 carbamate), the internal nucleophiles may close with a strong net preference for either syn or anti DOI: 10.1039/c7sc04430e addition relative to the Cl atom. Studies of both uncatalyzed and (DHQD)2PHAL-catalyzed -

The Reactions of Alkenes
The Reactions of Alkenes The Stereochemistry of Addition Reactions 1 Diverse Reactions of Alkenes Alkenes react with many electrophiles to give useful products by addition (often through special reagents) 2 Preparation of Alkenes: A Preview of Elimination Reactions • Alkenes are commonly made by – elimination of HX from alkyl halide (dehydrohalogenation) • Uses heat and KOH – elimination of H-OH from an alcohol (dehydration) • requires strong acids (sulfuric acid, 50 ºC) 3 A Regioselective Reaction A reaction in which one structural isomer is favored over another, leading to its predominance in the mixture of products. 4 A Stereoselective Reaction A reaction in which one stereoisomer in a mixture is produced more rapidly than another, resulting in predominance of the favored stereoisomer in the mixture of products. 5 A Stereospecific Reaction A reaction in which a particular stereoisomeric form of reactant gives one specific stereoisomer of product, while a different stereoisomeric form of reactant leads to a different single pure streoisomer of product. Stereospecific reaction is also stereoselective; however, stereoselective reaction is not stereospecific. 6 An Electrophilic Addition Reaction where HX = HF, HCl, HBr, and HI Reactivity of HF << HCl < HBr < HI since HF is less acidic and HI is most acidic. The rate of addition of HI is too fast to measure. 7 The Mechanism of the Reaction 8 Relative Stabilities of Carbocations 9 Hyperconjugation Stabilizes a Carbocation 10 The Difference in Carbocation Stability Determines the Products -

Development of New Domino Reactions of Alkylidene Meldrum's
Development of New Domino Reactions of Alkylidene Meldrum’s Acids Involving Friedel-Crafts Chemistry and Catalytic Conjugate Allylation of Alkylidene Meldrum’s Acids by Aaron Michael Dumas A thesis presented to the University of Waterloo in fulfilment of the thesis requirement for the degree of Doctor of Philosophy in Chemistry Waterloo, Ontario, Canada, 2009 © Aaron Michael Dumas 2009 I hereby declare that I am the sole author of this thesis. This is a true copy of my thesis, including any required final revisions, as accepted by my examiners. I understand that my thesis may be made electronically available to the public. ii Abstract Alkylidene Meldrum’s acids are very reactive acceptors in conjugate additions, and are known to be significantly more electrophilic than other α,β-unsaturated carbonyl electrophiles. They also offer advantages in terms of ease of preparation, purification and storage. Despite this, they are relatively underused in organic synthesis, and have been treated as something of a curiousity in the literature. The goal of my research was to demonstrate the utility of these molecules in new reactions that are not readily available to other electrophiles. To facilitate this work, new conditions for the Knoevenagel condensation of aldehydes with Meldrum’s acid were developed. This allowed access to a broader range of monosubstituted alkylidenes than was previously possible from any single method. In a reaction that exploits the acylating ability of Meldrum’s acid, a domino addition of phenols to alkylidene Meldrum’s acids was developed. Here, Yb(OTf)3 catalyzed the addition of a phenol to the alkylidene as well as acylation through activation of the electrophile. -

Chapter 8: Reactions of Alkanes; Radicals Page
REACTIONS OF ALKANES: CONTENTS Reactivity Considerations Chlorination and Bromination of Alkanes Reactivity–Selectivity Principle Radical Substitution of Benzylic and Allylic Hydrogens Stereochemistry of Radical Substitution No Radical Reactions in Biological Systems 2 RADICALS Sources include: Hydrogen peroxide . Alkyl peroxides . Light causes homolysis of the weak O-O bond . 3 HETEROLYSIS & HOMOLYSIS Heterolytic bond cleavage or heterolysis H Br H+ + Br Homolytic bond cleavage or homolysis H Br H + Br Homolysis produces radicals, which are very reactive species 4 RADICAL CHAIN REACTIONS Initiation turns stable species into radicals by breaking a bond. 5 RADICAL CHAIN REACTIONS Propagation causes products to form without resulting in net consumption of radicals. (production = consumption) 6 RADICAL CHAIN REACTIONS Termination results in net radical destruction. Generally occurs when reactants are used up. 7 CHLORINATION AND BROMINATION OF ALKANES C H A P T E R 12 8 PRODUCT DISTRIBUTION 9 CHLORINATION RATES 10 CHLORINATION PRODUCT DISTRIBUTION 11 BROMINATION RATES 12 REACTIVITY–SELECTIVITY PRINCIPLE The very reactive chlorine atom will have lower selectivity and attack pretty much any hydrogen available on an alkane The less reactive bromine atom will be more selective and tends to react preferentially with the easy targets, i.e. benzylic/allylic > 3° > 2° > 1° 13 RADICALS Stability of alkyl radicals is similar to stability of carbocations 14 CRUDE RADICAL STABILITY INDEX Add 1 for each attached carbon. Add 3 for adjacent double bond or phenyl ring. Radical equally stable on double bond carbon as on single bonded carbon Profile similar to carbocations but resonance contributes more to radical stability, and radical is OK on C=C. -
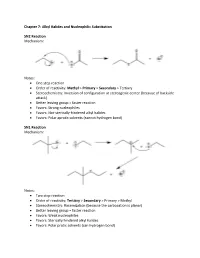
Alkyl Halides and Nucleophilic Substitution SN2 Reaction
Chapter 7: Alkyl Halides and Nucleophilic Substitution SN2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Methyl > Primary > Secondary > Tertiary • Stereochemistry: Inversion of configuration at stereogenic center (because of backside attack) • Better leaving group = faster reaction • Favors: Strong nucleophiles • Favors: Not-sterically-hindered alkyl halides • Favors: Polar aprotic solvents (cannot hydrogen bond) SN1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary > Methyl • Stereochemistry: Racemization (because the carbocation is planar) • Better leaving group = faster reaction • Favors: Weak nucleophiles • Favors: Sterically hindered alkyl halides • Favors: Polar protic solvents (can hydrogen bond) Important Trends Chapter 8: Alkyl Halides and Elimination Reactions E2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: antiperiplanar arrangement of H and X • Better leaving group = faster reaction • Favors: Polar aprotic solvents, strong bases • Products follow Zaitsev rule (more substituted alkene is the major product) E1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: Trigonal planar carbocation intermediate • Better leaving group = faster reaction • Favors: Polar protic solvents, weak bases • Products follow Zaitsev rule Chapter 9: Alcohols, Ethers, and Epoxides Preparation of Alcohols Mechanism: Notes: • SN2 mechanism -

Download (8MB)
THE MODIFICATION AND USE OF SUPPORTED PLATINUM CATALYSTS FOR ASYMMETRIC HYDROGENATION REACTIONS A Thesis Presented to die University of Glasgow for the Degree of Doctor of Philosophy by Elaine Allan October 1995 ProQuest Number: 11007860 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest 11007860 Published by ProQuest LLC(2018). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106- 1346 'fCiU l o z U CT 7 G la sg o w i UNIVERSITY ■ i t e y SUMMARY This thesis describes a study of the adsorption of chiral substituted binaphthalene molecules ( 2,2'-dihydroxy-l,l'-binaphthalene, 2,2'-diamino-l,l'-binaphthalene, 2,2- dimethoxy-1,1 '-binaphthalene and 2,2',7,7'-tetrahy droxy-1,1 ’-binaphthalene) on to supported Pt catalysts (1% w/w Pt/y-alumina, 1% w/w Pt/Grace silica CIO and 1% w/w Pt/Cab-O- Sil) with a view to establishing a system which could be capable of inducing asymmetric hydrogenation of prochiral starting materials. The 2,2'-dihydroxy-l,r-binaphthalene modifier was found to adsorb irreversibly on to the 1% w/w Pt/y-alumina and 1% w/w Pt/Grace silica CIO catalysts, prior to the ageing of the 1% w/w Pt/Grace silica CIO catalyst. -

Chemistry for Pharmacy Students : General, Organic
Chemistry for Pharmacy Students General, Organic and Natural Product Chemistry Satyajit D. Sarker University of Ulster, Coleraine, Northern Ireland, UK Lutfun Nahar University of Ulster, Coleraine, Northern Ireland, UK Chemistry for Pharmacy Students Chemistry for Pharmacy Students General, Organic and Natural Product Chemistry Satyajit D. Sarker University of Ulster, Coleraine, Northern Ireland, UK Lutfun Nahar University of Ulster, Coleraine, Northern Ireland, UK Copyright #2007 John Wiley & Sons Ltd, The Atrium, Southern Gate, Chichester, West Sussex PO19 8SQ, England Telephone (þ44) 1243 779777 Email (for orders and customer service enquiries): [email protected] Visit our Home Page on www.wiley.com All Rights Reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, mechanical, photocopying, recording, scanning or otherwise, except under the terms of the Copyright, Designs and Patents Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency Ltd, 90 Tottenham Court Road, London W1T 4LP, UK, without the permission in writing of the Publisher. Requests to the Publisher should be addressed to the Permissions Department, John Wiley & Sons Ltd, The Atrium, Southern Gate, Chichester, West Sussex PO19 8SQ, England, or emailed to [email protected], or faxed to (þ44) 1243 770620. Designations used by companies to distinguish their products are often claimed as trademarks. All brand names and product names used in this book are trade names, service marks, trademarks or registered trademarks of their respective owners. The Publisher is not associated with any product or vendor mentioned in this book. -

Alkynes Alkynes Or Acetylenes Are Compounds That Contain a Carbon–Carbon Triple Bond
Alkynes Alkynes or acetylenes are compounds that contain a carbon–carbon triple bond. E.g. H C C H CH3CH2 C C H H3C C C CH3 acetylene The triple bond results in a molecular formula of CnH2n-2 Ethane C2H6 0 elements of unsaturation Ethene C2H4 1 element of unsaturation Ethyne C2H2 2 elements of unsaturation The triple bond contributes two elements of unsaturation. Nomenclature of Alkynes IUPAC nomenclature is similar to that for alkenes, except the –ane ending is replaced with –yne. The chain is numbered from the end closest to the triple bond. Ch09 Alkynes (landscape).docx Page 1 When additional functional groups are present, the suffixes are combined. Terminal and Internal Alkynes The position of the triple bond can alter the reactivity of the alkyne. Compounds with triple bonds at the end of a molecule are called terminal alkynes. (Terminal C-H groups are called acetylenic hydrogens). If the triple bond is flanked by alkyl groups on both sides it is an internal alkyne. Ch09 Alkynes (landscape).docx Page 2 Electronic Structure of Alkynes The triple bond consists of one bond and two bonds. Each carbon is bonded to two other atoms, and there are no nonbonding electrons. Carbon requires two hybrid orbitals to bond to the atoms, thus sp hybrids are used. The sp orbitals are linear and oriented at 180°. The C-C bond is formed from sp-sp overlap. The C-H bond is formed from sp-s overlap. The creation of sp hybrids leaves 2 free p orbitals – these create the two bonds. -

Controlling the Geometry of Double Bonds 31 Connections
Controlling the geometry of double bonds 31 Connections Building on: Arriving at: Looking forward to: • Carbonyl chemistry ch6, ch12, & ch14 • What makes E- and Z-alkenes • Diastereoselectivity ch33–ch34 • Kinetic and thermodynamic control different? • Pericyclic reactions ch35–ch36 |ch13| • Why E/Z control matters • Fragmentations ch38 • Wittig reaction ch14 • Eliminations are not stereoselective • Radicals and carbenes ch39–ch40 • Conjugate addition ch10 • Cyclic alkenes are cis • Main group chemistry ch46–ch47 • Stereochemistry ch16 • Equilibration of alkenes gives trans • Asymmetric synthesis ch45 • Elimination reactions ch19 • Effects of light and how we see • Polymerization ch52 • Reduction ch24 • Julia olefination and the Wittig • Organic synthesis ch53 • Chemistry of enol(ate)s ch26–ch29 reaction at work • Reliable reduction of alkynes The properties of alkenes depend on their geometry You have met alkenes participating in reactions in a number of chapters, but our discussion of how to make alkenes has so far been quite limited. Chapter 19 was about elimination reactions, and there you met E1 and E2 reactions. OH OH2 E1 H3PO4 elimination H rate-determining step E2 Br OH Br elimination H OH rate-determining step In Chapter 14, you met an important reaction known as the Wittig reaction, which also forms alkenes. O Ph3PCH2 + Ph3PO Different physical properties: maleate and fumarate These two compounds, (Z)- and (E)-dimethyl but-2- can be. Dimethyl maleate is a liquid with a boiling point of enedioate, are commonly known as dimethyl maleate and 202 °C (it melts at –19 °C), while dimethyl fumarate is a dimethyl fumarate. They provide a telling example of how crystalline compound with a melting point of 103–104 °C. -

Difference Between Syn and Anti Addition Key Difference
Difference Between Syn and Anti Addition www.differencebetween.com Key Difference - Syn vs Anti Addition In organic chemistry, the addition reactions are characterized by two groups that bond to a double bond. During this characteristic addition reaction of alkenes, the p bond of the double bond is broken and new σ bonds are formed. It is because p bond of C=C bond is much weaker and unstable than C-C σ bond. Moreover, p bond of alkenes makes them electron rich, as p bond’s electron density is concentrated above and below the plane of the molecule. Therefore, p bond is more vulnerable to σ electrophiles than the bond. The stereochemistry is important in determining the mechanism of addition reactions. The stereochemistry of addition reactions depends on two aspects. First one is the joining side of electrophile and nucleophile to the double-bonded carbons (whether it is from the same side of the double bond or from the opposite side). The second aspect is the geometrical orientation of electrophile and nucleophile to each other and the rest of the organic molecule. Based on these aspects, there are two possible stereochemistries for the addition, syn and anti. The key difference between syn addition and anti addition is that in syn addition, both electrophile and nucleophile add from the same side of the plane of double-bonded carbon atoms, whereas in anti- addition, nucleophile and electrophile add from opposite sides of this plane. Further details related to syn and anti additions are discussed below. What is Syn Addition? Syn addition is a possible stereochemistry of addition where both electrophile and nucleophile bond to the same side of the plane of the double-bonded carbon atoms of an alkene. -
CHEM 263 (AS 02) Organic Chemistry II Winter 2020 Instructor:Dr.Manzar Saberi Office: 5- 138 G, City Center Campus Phone: (780) 497-4634 Email: [email protected]
CHEM 263 (AS 02) Organic Chemistry II Winter 2020 Instructor:Dr.Manzar Saberi Office: 5- 138 G, City Center Campus Phone: (780) 497-4634 Email: [email protected] Lectures: Tuesday, Thursday11:00-12:20CCC Room 5-152 Office Hours:Tuesday, Thursday 9:00-11:00 Total Course Hours: Lecture 45 hours, Laboratory 36 hours Web address:http://academic.macewan.ca/saberim Textbook: Organic Chemistry by T. W. Graham Solomons and Craig B. Fryhle,12thedition Laboratory Manual: Organic Chemistry 263 Laboratory Manual edition 2019/20 Optional Materials: Study Guide and Solution manualand molecular model kit. Grade Distribution: Term I Exam- Thursday, February 6(80 min) 22.5 % Term II Exam- Thursday March 12(80 min) 22.5 % Final Examination (3.0 Hours) 30 % Laboratory Component 25% Laboratory Examination: March 31st 6:00-7:30 PM Note: Students are responsible for verifying the date of the final exam (see here). Course Description: (3 credits)The nomenclature, structure, physical properties, synthesis and selected reactions of the basic functional groups in organic chemistry are discussed. Functional groups covered include alkenes, alkynes, aromatic compounds, alcohols, phenols, ethers, aldehydes, ketones, amines, carboxylic acids and carboxylic acid derivatives. The presence of these functional groups in natural products is emphasized. The application of spectroscopic methods for structure determination in simple organic molecules is discussed. Prerequisite: Minimum grade of C- in CHEM 164 or 261 Laboratory: Laboratory classes begin the second week of the term. The laboratory component is compulsory for credit in CHEM 263; attendance is mandatory and no make-up labs are available. If you know that you will be unable to attend a scheduled laboratory period, it is your responsibility to inform your laboratory instructor at least one week prior so that you can complete the experiment in another laboratory session. -

Alkynes Alkynes Or Acetylenes Are Compounds That Contain a Carbon–Carbon Triple Bond
Alkynes Alkynes or acetylenes are compounds that contain a carbon–carbon triple bond. E.g. H C C H CH3CH2 C C H H3C C C CH3 acetylene The triple bond results in a molecular formula of CnH2n-2 Ethane C2H6 0 elements of unsaturation Ethene C2H4 1 element of unsaturation Ethyne C2H2 2 elements of unsaturation The triple bond contributes two elements of unsaturation. Nomenclature of Alkynes IUPAC nomenclature is similar to that for alkenes, except the –ane ending is replaced with –yne. The chain is numbered from the end closest to the triple bond. H H H C C H H3C C C CH3 H3C C C C CH2 C CH3 CH Br ethyne 2-butyne 3 6-bromo-2-methyl-3-heptyne but-2-yne 6-bromo-2-methylhept-3-yne When additional functional groups are present, the suffixes are combined. H H H2C C C C CH3 H3C C C C H H3C C C C CH2CH3 CH3 OH OCH3 2-methyl-1-penten-3-yne 3-butyn-2-ol 4-methoxy-2-hexyne Ch09Su13 2013 Page1 Terminal and Internal Alkynes The position of the triple bond can alter the reactivity of the alkyne. Compounds with triple bonds at the end of a molecule are called terminal alkynes. (Terminal C-H groups are called acetylenic hydrogens). H3C C C H H3C C C CH3 terminal internal alkyne alkyne If the triple bond is flanked by alkyl groups on both sides it is an internal alkyne. Electronic Structure of Alkynes The triple bond consists of one bond and two bonds.