Controlling the Geometry of Double Bonds 31 Connections
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Absolute and Relative Facial Selectivities in Organocatalytic Asymmetric Chlorocyclization Reactions
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Absolute and relative facial selectivities in organocatalytic asymmetric chlorocyclization Cite this: Chem. Sci.,2018,9, 2898 reactions† Nastaran Salehi Marzijarani,a Roozbeh Yousefi,a Arvind Jaganathan,b Kumar Dilip Ashtekar,a James E. Jackson *a and Babak Borhan *a Though (DHQD)2PHAL-catalyzed chlorocyclizations of 1,1-disubstituted olefins show useful (and in some cases, reversible) asymmetric induction, stereochemically complete descriptions of these alkene additions have remained largely unknown. Herein, based on a combination of NMR, derivative, isotope labeling, and computational studies, we present detailed stereochemical analyses of chlorocyclizations of nucleophile-tethered 1,1-disubstituted styryl systems. The selectivities of the two asymmetric bond- forming processes, namely electrophilic chlorine attack and nucleophilic ring closure, are thus mapped out independently. Under the established optimal conditions, four related chlorocyclizations were + Creative Commons Attribution 3.0 Unported Licence. subjected to this analysis. All showed a strong preference for Cl delivery from the same face of the Received 12th October 2017 alkene. However, depending on reaction conditions and substrate identity (carboxylic acid, amide or Accepted 24th December 2017 carbamate), the internal nucleophiles may close with a strong net preference for either syn or anti DOI: 10.1039/c7sc04430e addition relative to the Cl atom. Studies of both uncatalyzed and (DHQD)2PHAL-catalyzed -

The Reactions of Alkenes
The Reactions of Alkenes The Stereochemistry of Addition Reactions 1 Diverse Reactions of Alkenes Alkenes react with many electrophiles to give useful products by addition (often through special reagents) 2 Preparation of Alkenes: A Preview of Elimination Reactions • Alkenes are commonly made by – elimination of HX from alkyl halide (dehydrohalogenation) • Uses heat and KOH – elimination of H-OH from an alcohol (dehydration) • requires strong acids (sulfuric acid, 50 ºC) 3 A Regioselective Reaction A reaction in which one structural isomer is favored over another, leading to its predominance in the mixture of products. 4 A Stereoselective Reaction A reaction in which one stereoisomer in a mixture is produced more rapidly than another, resulting in predominance of the favored stereoisomer in the mixture of products. 5 A Stereospecific Reaction A reaction in which a particular stereoisomeric form of reactant gives one specific stereoisomer of product, while a different stereoisomeric form of reactant leads to a different single pure streoisomer of product. Stereospecific reaction is also stereoselective; however, stereoselective reaction is not stereospecific. 6 An Electrophilic Addition Reaction where HX = HF, HCl, HBr, and HI Reactivity of HF << HCl < HBr < HI since HF is less acidic and HI is most acidic. The rate of addition of HI is too fast to measure. 7 The Mechanism of the Reaction 8 Relative Stabilities of Carbocations 9 Hyperconjugation Stabilizes a Carbocation 10 The Difference in Carbocation Stability Determines the Products -

Selected Reactions of Thiocarbonyl Compounds Oac O Ncy Oac CN 2 Shyam Krishnan Me Meo Monday, June 12, 2006 Me H Me N O H 8 P.M
OH S Me S O O N SEt TBSO Ph O Me Me N OH BnO S Selected Reactions of Thiocarbonyl Compounds OAc O NCy OAc CN 2 Shyam Krishnan Me MeO Monday, June 12, 2006 Me H Me N O H 8 p.m. N O Me 147 Noyes H O O O HN O H H S Me S S H H BnO BnO N N i) ICH2CO2Et, CHCl3 S ii) PPh , DABCO, 3 CO2Et CHCl3, reflux (92% yield) I I OMe OMe Selected Reactions of Thiocarbonyl Compounds 1) Thiocarbonyl compounds: nomenclature and structural properties 2) Methods of Synthesis 3) Reactions of thiocarbonyl compounds and their application in the synthesis of functionalized molecules 1) Reactions of carbanions derived from Thiocarbonyl compounds. 2) Carbanion addition to the thiocarbonyl group. 3) Reactions with electrophiles - the Eschenmoser sulfide contraction. 4) Radical mediated reactions. 5) [3,3] sigmatropic rearrangements - the thio-Claisen rearrangement. 6) [4+2] cycloaddition reactions. 7) [3+2] Dipolar cycloadditions. 8) Summary and future directions. Reviews: General review: Metzner, P. Top. Curr. Chem. 1999, 204, 127. General review: Metzner, P. Synthesis 1992, 1185. Synthesis of heterocycles: Jagodzinski, T.S. Chem. Rev. 2003, 103, 197. Radical chemistry: Crich, D.; Quintero, L. Chem. Rev. 1989, 89, 1413. Photochemistry: Coyle, J. D. Tetrahedron 1985, 41, 5393. Thiocarbonyl Compounds Structures, Nomenclature and Stability Thiocarbonyl compounds possess a carbon-sulfur double bond Thiocarbonyl compounds with at least one organic group bound to the thiocarbonyl carbon: O S S S S S S R H R R' R OR R NR2 R SR R R' Thioaldehyde Thioketone Thionoester Thioamide Dithioester Sulfine/Thiocarbonyl oxide Typically display greater reactivity than their carbonyl (oxygen) analogs • Larger covalent radius of sulfur vs oxygen (104.9 nm vs 70.2 nm), less efficient overlap in S3p-C2p π-bond • Dissociation energy of C=S (115 kcal/mol) is significantly lower than for C=O (162 kcal/mol). -

Development of New Domino Reactions of Alkylidene Meldrum's
Development of New Domino Reactions of Alkylidene Meldrum’s Acids Involving Friedel-Crafts Chemistry and Catalytic Conjugate Allylation of Alkylidene Meldrum’s Acids by Aaron Michael Dumas A thesis presented to the University of Waterloo in fulfilment of the thesis requirement for the degree of Doctor of Philosophy in Chemistry Waterloo, Ontario, Canada, 2009 © Aaron Michael Dumas 2009 I hereby declare that I am the sole author of this thesis. This is a true copy of my thesis, including any required final revisions, as accepted by my examiners. I understand that my thesis may be made electronically available to the public. ii Abstract Alkylidene Meldrum’s acids are very reactive acceptors in conjugate additions, and are known to be significantly more electrophilic than other α,β-unsaturated carbonyl electrophiles. They also offer advantages in terms of ease of preparation, purification and storage. Despite this, they are relatively underused in organic synthesis, and have been treated as something of a curiousity in the literature. The goal of my research was to demonstrate the utility of these molecules in new reactions that are not readily available to other electrophiles. To facilitate this work, new conditions for the Knoevenagel condensation of aldehydes with Meldrum’s acid were developed. This allowed access to a broader range of monosubstituted alkylidenes than was previously possible from any single method. In a reaction that exploits the acylating ability of Meldrum’s acid, a domino addition of phenols to alkylidene Meldrum’s acids was developed. Here, Yb(OTf)3 catalyzed the addition of a phenol to the alkylidene as well as acylation through activation of the electrophile. -

Total Synthesis of (+)-Leinamycin
Total Synthesis of (+)-Leinamycin Tohru Fukuyama *,•õ and Yutaka Kanda •õ †Department of Chemistry, Rice University, TTokyo Research Laboratories, Kyowa Hakko Kogyo Co .,Ltd., Abstract : A total synthesis of (+)-leinamycin (I), a unique sulfur-containing antitumor antibiotic, is described. The present synthesis features a stereocontrolled construction of the macrolactam followed by a spiroannulation of the hitherto unprecedented dithiolanone ring. 1. Introduction Leinamycin (1) has recently been isolated from a fermentation broth of Streptomyces sp. by a Kyowa Hakko group and has been shown to exhibit potent antitumor activities against experimental tumors (ref. 1). Leinamycin preferentially inhibits DNA synthesis in Bacillus subtilis and causes single strand scission of plasmid DNA in vitro in the presence of thiol cofactors (ref. 2). The relative configuration of leinamycin was determined by an X-ray crystallographic analysis (ref. 3). The absolute configuration was deduced as shown on the basis of the fact that partially racemized D-alanine was obtained by acid hydrolysis of leinamycin (ref. 4). The unique structural features of leinamycin include the 1,3-dioxo-1,2-dithiolane moiety which is fused in a spiro fashion to an 18-membered lactam and the extensively conjugated thiazole ring. No other natural products with such an unusual dithiolanone moiety have been isolated to date. These synthetically challenging structural features combined with the interesting antitumor activities prompted us to initiate synthetic studies on leinamycin at the beginning of 1989. Completion of our first total synthesis of leinamycin was achieved at the end of 1992 and has recently been reported as a communication (ref. 5, 6). Leinamycin 2. -

United States Patent (19) 11 Patent Number: 6,051,704 Gordon-Wylie Et Al
US006051704A United States Patent (19) 11 Patent Number: 6,051,704 Gordon-Wylie et al. (45) Date of Patent: Apr. 18, 2000 54 SYNTHESIS OF MACROCYCLIC Erich Stuart Uffelman, Macrocyclic Tetraamido-N Ligands TETRAAMIDO-N LIGANDS that Stabilize High Valent Complexes of Chromium, Maganese, Iron, Cobalt, Nickel and Copper, California 75 Inventors: Scott W. Gordon-Wylie; Terrence J. Institute of Technology, Aug. 19, 1991. Collins, both of Pittsburgh, Pa. Theodora W. Greene, Protective Groups in Organic Synthe sis, Harvard University, John Wiley & Sons, 1981. 73 Assignee: Carnegie Mellon University, Kimberely K. Kostka, Synthesis and Characterization of Pittsburgh, Pa. High-Valent Iron Complexes of Macrocyclic Tetraamido-N Ligands, Carnegie Mellon University, Jul. 19, 1993. 21 Appl. No.: 08/681,187 Nathan L. Drake, Harry D. Anspon, et al. Synthetic Anti marlarials. Some Derivatives of 8-Aminoquinoline, Labo 22 Filed: Jul. 22, 1996 ratories of the University of Maryland, vol. 68, p. 1536, Aug. 51) Int. Cl." ....................... C07D 403/02; CO7D 259/00 1946. 52 U.S. Cl. .......................... 540/465; 540/451; 540/460; Richard J. Bushby and Michael D. Pollard, The Introduction of Alkylidene Substituents into the 4-Position of the 3,3,5, 540/463; 540/450 5, Tetramethyl-A-pyrazoline Nucleus by the Thioketone 58 Field of Search ..................................... 540/450, 451, plus Diazoalkane Reaction: Synthesis of Tetrasubstituted 540/460, 452, 453, 465, 463 Episulphides and Alkenes. 56) References Cited Primary Examiner Mukund J. Shah Assistant Examiner Pavanaram K. Sripada U.S. PATENT DOCUMENTS Attorney, Agent, or Firm Kirkpatrick & Lockhart LLP 4,517,122 5/1985 Tomalia et al. ...................... 260/239.3 57 ABSTRACT 4,577,042 3/1986 Collins et al. -

Chapter 8: Reactions of Alkanes; Radicals Page
REACTIONS OF ALKANES: CONTENTS Reactivity Considerations Chlorination and Bromination of Alkanes Reactivity–Selectivity Principle Radical Substitution of Benzylic and Allylic Hydrogens Stereochemistry of Radical Substitution No Radical Reactions in Biological Systems 2 RADICALS Sources include: Hydrogen peroxide . Alkyl peroxides . Light causes homolysis of the weak O-O bond . 3 HETEROLYSIS & HOMOLYSIS Heterolytic bond cleavage or heterolysis H Br H+ + Br Homolytic bond cleavage or homolysis H Br H + Br Homolysis produces radicals, which are very reactive species 4 RADICAL CHAIN REACTIONS Initiation turns stable species into radicals by breaking a bond. 5 RADICAL CHAIN REACTIONS Propagation causes products to form without resulting in net consumption of radicals. (production = consumption) 6 RADICAL CHAIN REACTIONS Termination results in net radical destruction. Generally occurs when reactants are used up. 7 CHLORINATION AND BROMINATION OF ALKANES C H A P T E R 12 8 PRODUCT DISTRIBUTION 9 CHLORINATION RATES 10 CHLORINATION PRODUCT DISTRIBUTION 11 BROMINATION RATES 12 REACTIVITY–SELECTIVITY PRINCIPLE The very reactive chlorine atom will have lower selectivity and attack pretty much any hydrogen available on an alkane The less reactive bromine atom will be more selective and tends to react preferentially with the easy targets, i.e. benzylic/allylic > 3° > 2° > 1° 13 RADICALS Stability of alkyl radicals is similar to stability of carbocations 14 CRUDE RADICAL STABILITY INDEX Add 1 for each attached carbon. Add 3 for adjacent double bond or phenyl ring. Radical equally stable on double bond carbon as on single bonded carbon Profile similar to carbocations but resonance contributes more to radical stability, and radical is OK on C=C. -
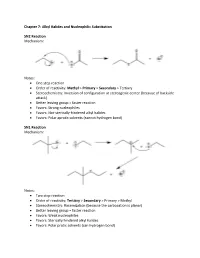
Alkyl Halides and Nucleophilic Substitution SN2 Reaction
Chapter 7: Alkyl Halides and Nucleophilic Substitution SN2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Methyl > Primary > Secondary > Tertiary • Stereochemistry: Inversion of configuration at stereogenic center (because of backside attack) • Better leaving group = faster reaction • Favors: Strong nucleophiles • Favors: Not-sterically-hindered alkyl halides • Favors: Polar aprotic solvents (cannot hydrogen bond) SN1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary > Methyl • Stereochemistry: Racemization (because the carbocation is planar) • Better leaving group = faster reaction • Favors: Weak nucleophiles • Favors: Sterically hindered alkyl halides • Favors: Polar protic solvents (can hydrogen bond) Important Trends Chapter 8: Alkyl Halides and Elimination Reactions E2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: antiperiplanar arrangement of H and X • Better leaving group = faster reaction • Favors: Polar aprotic solvents, strong bases • Products follow Zaitsev rule (more substituted alkene is the major product) E1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: Trigonal planar carbocation intermediate • Better leaving group = faster reaction • Favors: Polar protic solvents, weak bases • Products follow Zaitsev rule Chapter 9: Alcohols, Ethers, and Epoxides Preparation of Alcohols Mechanism: Notes: • SN2 mechanism -

Download (8MB)
THE MODIFICATION AND USE OF SUPPORTED PLATINUM CATALYSTS FOR ASYMMETRIC HYDROGENATION REACTIONS A Thesis Presented to die University of Glasgow for the Degree of Doctor of Philosophy by Elaine Allan October 1995 ProQuest Number: 11007860 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest 11007860 Published by ProQuest LLC(2018). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106- 1346 'fCiU l o z U CT 7 G la sg o w i UNIVERSITY ■ i t e y SUMMARY This thesis describes a study of the adsorption of chiral substituted binaphthalene molecules ( 2,2'-dihydroxy-l,l'-binaphthalene, 2,2'-diamino-l,l'-binaphthalene, 2,2- dimethoxy-1,1 '-binaphthalene and 2,2',7,7'-tetrahy droxy-1,1 ’-binaphthalene) on to supported Pt catalysts (1% w/w Pt/y-alumina, 1% w/w Pt/Grace silica CIO and 1% w/w Pt/Cab-O- Sil) with a view to establishing a system which could be capable of inducing asymmetric hydrogenation of prochiral starting materials. The 2,2'-dihydroxy-l,r-binaphthalene modifier was found to adsorb irreversibly on to the 1% w/w Pt/y-alumina and 1% w/w Pt/Grace silica CIO catalysts, prior to the ageing of the 1% w/w Pt/Grace silica CIO catalyst. -

Regioselectivity in the Trimethylsilyl Thioketone with 1,3-Butadienes
Regioselectivity in the Reactions of Thioacylsilane with Dienes Bull. Korean Chem. Soc., V이 . 13, No. 1, 1992 41 Chem., 20, 445 (1981). 26. T. Bryant and D. L. H. Williams, J. Chem. Soc., Perkin 23. K. A. Jorgensen and S.-O. Lawesson, J. Am. Chem. Soc., Trans. 2, 97 (1988). 106, 4687 (1984). 27. P. A. Morris and D. L. H. Williams, J. Chem. Soc.t Perkin 24. M. R. Crampton, J. T. Thomson, and D. L. H. Williams, Trans. 2, 513 (1988). / Chem. Soc., Perkin Trans. 2, 18 (1979). 28. T. A. Meyer and D. L. H. Williams, J. Chem. Soc.t Perkin 25. A. Castro, J. R. Leis, and M. E. Pena, J. Chem. Res. (S), Trans. 2, 517 (1988). 216 (1986). Regioselectivity in the Cycloaddition Reactions of t-Butyl Trimethylsilyl Thioketone with 1,3-Butadienes Kyung-Tae Kang*, Chi Hyo Park, and Ung Chan Yoonf Department of Chemical Education and ^Department of Chemistry, Pusan National University, Pusan 609-735. Received August 22, 1991 Thermal cycloaddition of /-butyl trimethylsilyl thioketone (1) with 2-substituted dienes such as isoprene and 2-trimethyl- silyloxy-l,3-butadiene occurred smoothly at 80M to afford regioiomeric mixtures of cycloadducts. On the other hand, similar treatment of 1 with 1-substituted dienes such as fraws-l.S-pentadiene, 1-methoxy- and l-acetoxy-lt3-butadiene and Danishefsky's diene afforded a single regioisomeric adduct, respectively. Protodesilylation of the silylated adducts 8 and 11 could also be performed with ease. Introduction The cycloadducts were identified to be 5-methyl-2-Z-bu- tyl-2-trimethylsilyl-3,6-dihydro-2Af-thiopyran 2a and 4-methyl Thioacylsilanes have received increasing attention in re analog 2b on the basis of spectral data. -

Facile and Efficient Synthesis of Thiosemicarbazone Derivatives with Functionalized Pendant Amines
University of Louisville ThinkIR: The University of Louisville's Institutional Repository College of Arts & Sciences Senior Honors Theses College of Arts & Sciences 3-2019 Facile and efficient synthesis of thiosemicarbazone derivatives with functionalized pendant amines. Anna E Davis University of Louisville Follow this and additional works at: https://ir.library.louisville.edu/honors Part of the Inorganic Chemistry Commons, and the Organic Chemistry Commons Recommended Citation Davis, Anna E, "Facile and efficient synthesis of thiosemicarbazone derivatives with functionalized pendant amines." (2019). College of Arts & Sciences Senior Honors Theses. Paper 202. Retrieved from https://ir.library.louisville.edu/honors/202 This Senior Honors Thesis is brought to you for free and open access by the College of Arts & Sciences at ThinkIR: The nivU ersity of Louisville's Institutional Repository. It has been accepted for inclusion in College of Arts & Sciences Senior Honors Theses by an authorized administrator of ThinkIR: The nivU ersity of Louisville's Institutional Repository. This title appears here courtesy of the author, who has retained all other copyrights. For more information, please contact [email protected]. Facile and Efficient Synthesis of Thiosemicarbazone Derivatives with Functionalized Pendant Amines. By Anna Elizabeth Davis Submitted in partial fulfillment of the requirements for Graduation summa cum laude and for Graduation with Honors from the Department of Chemistry University of Louisville May, 2019 1 Facile and efficient -

Sulfur-Facilitated Organic Synthesis
Sulfur-Facilitated Organic Synthesis Andrew McClory Monday March 16, 2009 8:00 pm, 147 Noyes S S S S S S 'Attempts to make thioacetone by the cracking of trithioacetone gave rise to an offensive smell which spread rapidly over a great area of the town causing fainting, vomiting and a panic evacuation'...'the laboratory work was abandoned.' -Researcher, Freiburg, 1889 S S S 'Attempts to make thioacetone by the cracking of trithioacetone gave rise to an offensive smell which spread rapidly over a great area of the town causing fainting, vomiting and a panic evacuation'...'the laboratory work was abandoned.' -Researcher, Freiburg, 1889 'Recently we found ourselves with an odour problem beyond our worst expectations. During early experiments, a stopper jumped from a bottle of residues, and, although replaced at once, resulted in an immediate complaint of nausea and sickness from colleagues working in a building two hundred yards away. Two of our chemists who had done no more than investigate the cracking of minute amounts of trithioacetone found themselves the object of hostile stares in a restaurant and suffered the humiliation of having a waitress spray the area around them with a deodorant. The odours defied the expected effects of dilution since workers in the laboratory did not find the odours intolerable...and genuinely denied responsibility since they were working in closed systems. To convince them otherwise, they were dispersed with other observers around the laboratory, at distances up to a quarter of a mile, and one drop of either acetone gem-dithiol or the mother liquors from crude thioacetone crystallizations were placed on a watch glass in a fume cupboard.