Alcohols, Ethers, and Epoxides
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 21 the Chemistry of Carboxylic Acid Derivatives
Instructor Supplemental Solutions to Problems © 2010 Roberts and Company Publishers Chapter 21 The Chemistry of Carboxylic Acid Derivatives Solutions to In-Text Problems 21.1 (b) (d) (e) (h) 21.2 (a) butanenitrile (common: butyronitrile) (c) isopentyl 3-methylbutanoate (common: isoamyl isovalerate) The isoamyl group is the same as an isopentyl or 3-methylbutyl group: (d) N,N-dimethylbenzamide 21.3 The E and Z conformations of N-acetylproline: 21.5 As shown by the data above the problem, a carboxylic acid has a higher boiling point than an ester because it can both donate and accept hydrogen bonds within its liquid state; hydrogen bonding does not occur in the ester. Consequently, pentanoic acid (valeric acid) has a higher boiling point than methyl butanoate. Here are the actual data: INSTRUCTOR SUPPLEMENTAL SOLUTIONS TO PROBLEMS • CHAPTER 21 2 21.7 (a) The carbonyl absorption of the ester occurs at higher frequency, and only the carboxylic acid has the characteristic strong, broad O—H stretching absorption in 2400–3600 cm–1 region. (d) In N-methylpropanamide, the N-methyl group is a doublet at about d 3. N-Ethylacetamide has no doublet resonances. In N-methylpropanamide, the a-protons are a quartet near d 2.5. In N-ethylacetamide, the a- protons are a singlet at d 2. The NMR spectrum of N-methylpropanamide has no singlets. 21.9 (a) The first ester is more basic because its conjugate acid is stabilized not only by resonance interaction with the ester oxygen, but also by resonance interaction with the double bond; that is, the conjugate acid of the first ester has one more important resonance structure than the conjugate acid of the second. -

Absolute and Relative Facial Selectivities in Organocatalytic Asymmetric Chlorocyclization Reactions
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Absolute and relative facial selectivities in organocatalytic asymmetric chlorocyclization Cite this: Chem. Sci.,2018,9, 2898 reactions† Nastaran Salehi Marzijarani,a Roozbeh Yousefi,a Arvind Jaganathan,b Kumar Dilip Ashtekar,a James E. Jackson *a and Babak Borhan *a Though (DHQD)2PHAL-catalyzed chlorocyclizations of 1,1-disubstituted olefins show useful (and in some cases, reversible) asymmetric induction, stereochemically complete descriptions of these alkene additions have remained largely unknown. Herein, based on a combination of NMR, derivative, isotope labeling, and computational studies, we present detailed stereochemical analyses of chlorocyclizations of nucleophile-tethered 1,1-disubstituted styryl systems. The selectivities of the two asymmetric bond- forming processes, namely electrophilic chlorine attack and nucleophilic ring closure, are thus mapped out independently. Under the established optimal conditions, four related chlorocyclizations were + Creative Commons Attribution 3.0 Unported Licence. subjected to this analysis. All showed a strong preference for Cl delivery from the same face of the Received 12th October 2017 alkene. However, depending on reaction conditions and substrate identity (carboxylic acid, amide or Accepted 24th December 2017 carbamate), the internal nucleophiles may close with a strong net preference for either syn or anti DOI: 10.1039/c7sc04430e addition relative to the Cl atom. Studies of both uncatalyzed and (DHQD)2PHAL-catalyzed -

The Reactions of Alkenes
The Reactions of Alkenes The Stereochemistry of Addition Reactions 1 Diverse Reactions of Alkenes Alkenes react with many electrophiles to give useful products by addition (often through special reagents) 2 Preparation of Alkenes: A Preview of Elimination Reactions • Alkenes are commonly made by – elimination of HX from alkyl halide (dehydrohalogenation) • Uses heat and KOH – elimination of H-OH from an alcohol (dehydration) • requires strong acids (sulfuric acid, 50 ºC) 3 A Regioselective Reaction A reaction in which one structural isomer is favored over another, leading to its predominance in the mixture of products. 4 A Stereoselective Reaction A reaction in which one stereoisomer in a mixture is produced more rapidly than another, resulting in predominance of the favored stereoisomer in the mixture of products. 5 A Stereospecific Reaction A reaction in which a particular stereoisomeric form of reactant gives one specific stereoisomer of product, while a different stereoisomeric form of reactant leads to a different single pure streoisomer of product. Stereospecific reaction is also stereoselective; however, stereoselective reaction is not stereospecific. 6 An Electrophilic Addition Reaction where HX = HF, HCl, HBr, and HI Reactivity of HF << HCl < HBr < HI since HF is less acidic and HI is most acidic. The rate of addition of HI is too fast to measure. 7 The Mechanism of the Reaction 8 Relative Stabilities of Carbocations 9 Hyperconjugation Stabilizes a Carbocation 10 The Difference in Carbocation Stability Determines the Products -

United States Patent 15) 3,669,956 Borck Et Al
United States Patent 15) 3,669,956 Borck et al. (45) June 13, 1972 54) 4-SUBSTITUTEDAMNO 260/472, 260/516, 260/518 R, 260/518 A, 260/519, PHENYACETIC ACDS AND 260/556 AR, 260/556 B, 260/558 S, 260/558 A, DERVATIVES THEREOF 260/559 T, 260/559 A, 260/.571, 260/574, 260/.575, (72 Inventors: Joachim Borck; Johann Dahin; Volker 424/244, 424/246, 424/248, 424/250, 424/267, Koppe; Josef Kramer; Gustav Shorre; J. 424/270, 424/272, 424/273, 424/274, 424/304, W. Hermann Hovy; Ernst Schorscher, all 424/309, 424/32 i, 424/324, 424/330 of Darmstadt, Germany 51) int. Cl. ........................................................ C07d 41/04 58) Field of Search........ 260/294X,293.4, 293.47, 239 BF, 73) Assignee: E. Merck A. G., Darmstadt, Germany 260/326.3, 294.3 E (22) Filed: July 22, 1968 56) References Cited (21) Appl. No.: 746,326 UNITED STATES PATENTS (30) Foreign Application Priority Data 3,252,970 5/1966 Huebner................................ 260/239 July 22, 1967 Germany.............................. M 74881 3,385,852 5/1968 Casadio................................. 260/246 Jan. 8, 1968 Germany... ....M 76850 OTHER PUBLICATIONS Feb. 23, 1968 Germany...... ...M 77363 March 1, 1968 Germany.............................. M 77429 Norman et al., J. Chen. Soc. 1963, (Nov.), 5431-6. (52) U.S. Cl................... 260/239 BF, 260/239 A, 260/239 E, Primary Examiner-Henry R. Jiles 260/243 B, 260/246, 260/247. 1, 260/247.2 R, Assistant Examiner-G. Thomas Todd 260/247.2 A, 260/247.2 B, 260/247.5 R, 260/247.7 Attorney-Millen, Raptes & White A, 260/247.7 H, -

Functionalized Hybrid Silicones – Catalysis, Synthesis and Application
Technische Universität München Fakultät für Chemie Fachgebiet Molekulare Katalyse Functionalized Hybrid Silicones – Catalysis, Synthesis and Application Sophie Luise Miriam Putzien Vollständiger Abdruck der von der Fakultät für Chemie der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) genehmigten Dissertation. Vorsitzender: Univ.-Prof. Dr. M. Schuster Prüfer der Dissertation: 1. Univ.-Prof. Dr. F. E. Kühn 2. Univ.-Prof. Dr. O.Nuyken (i.R.) Die Dissertation wurde am 16.02.2012 bei der Technischen Universität München eingereicht und durch die Fakultät für Chemie am 08.03.2012 angenommen. The following dissertation was prepared between April 2009 and March 2012 at the Chair of Inorganic Chemistry, Department of Molecular Catalysis of the Technische Universität München. I would like to express my deep gratitude to my academic supervisor Prof. Dr. Fritz E. Kühn for his support and confidence and the freedom of scientific research. This work was supported by a research grant from the BASF Construction Chemicals GmbH, Trostberg, Germany. Acknowledgement I would like to express my sincere gratitude to Prof. Dr. Oskar Nuyken and Dr. Eckhart Louis for their ongoing support and their undamped enthusiasm for my research topic. They supported this work with many inspiring discussions, new ideas and critical questions. I thank the BASF Construction Chemicals GmbH, Trostberg, for giving me the opportunity to work on an industrial cooperation project. Especially, I would like to thank Dr. Simone Klapdohr and Dr. Burkhard Walther, who accompanied this project from the industrial perspectice, for their support and the nice time I had in Trostberg during the application technological tests. -

Understanding the Thermochemical Conversion of Biomass to Overcome Biomass Recalcitrance Kwang Ho Kim Iowa State University
Iowa State University Capstones, Theses and Graduate Theses and Dissertations Dissertations 2015 Understanding the thermochemical conversion of biomass to overcome biomass recalcitrance Kwang Ho Kim Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/etd Part of the Agriculture Commons, and the Bioresource and Agricultural Engineering Commons Recommended Citation Kim, Kwang Ho, "Understanding the thermochemical conversion of biomass to overcome biomass recalcitrance" (2015). Graduate Theses and Dissertations. 14382. https://lib.dr.iastate.edu/etd/14382 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Understanding the thermochemical conversion of biomass to overcome biomass recalcitrance by Kwang Ho Kim A dissertation submitted to the graduate faculty in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Major: Agricultural and Biosystems Engineering Program of Study Committee: Robert C. Brown, Co-Major Professor Xianglan Bai, Co-Major Professor Kurt Rosentrater Matthew Darr Brent Shanks Young Jin Lee Iowa State University Ames, Iowa 2015 Copyright © Kwang Ho Kim, 2015. All rights reserved. ii DEDICATION Dedicated to my family for their unwavering support -

Development of New Domino Reactions of Alkylidene Meldrum's
Development of New Domino Reactions of Alkylidene Meldrum’s Acids Involving Friedel-Crafts Chemistry and Catalytic Conjugate Allylation of Alkylidene Meldrum’s Acids by Aaron Michael Dumas A thesis presented to the University of Waterloo in fulfilment of the thesis requirement for the degree of Doctor of Philosophy in Chemistry Waterloo, Ontario, Canada, 2009 © Aaron Michael Dumas 2009 I hereby declare that I am the sole author of this thesis. This is a true copy of my thesis, including any required final revisions, as accepted by my examiners. I understand that my thesis may be made electronically available to the public. ii Abstract Alkylidene Meldrum’s acids are very reactive acceptors in conjugate additions, and are known to be significantly more electrophilic than other α,β-unsaturated carbonyl electrophiles. They also offer advantages in terms of ease of preparation, purification and storage. Despite this, they are relatively underused in organic synthesis, and have been treated as something of a curiousity in the literature. The goal of my research was to demonstrate the utility of these molecules in new reactions that are not readily available to other electrophiles. To facilitate this work, new conditions for the Knoevenagel condensation of aldehydes with Meldrum’s acid were developed. This allowed access to a broader range of monosubstituted alkylidenes than was previously possible from any single method. In a reaction that exploits the acylating ability of Meldrum’s acid, a domino addition of phenols to alkylidene Meldrum’s acids was developed. Here, Yb(OTf)3 catalyzed the addition of a phenol to the alkylidene as well as acylation through activation of the electrophile. -

Chapter 18 Ethers and Epoxides; Thiols and Sulfides Ethers
Chapter 18 Ethers and Epoxides; Thiols and Sulfides Ethers • Ethers (R–O–R’): – Organic derivatives of water, having two organic groups bonded to the same oxygen atom © 2016 Cengage Learning 2 NAMES AND PROPERTIES OF ETHERS 3 Nomenclature: Common Names • Simple ethers are named by identifying two organic substituents and adding the word ether – Name the groups in alphabetical order – Symmetrical: Use dialkyl or just alkyl © 2016 Cengage Learning 4 Nomenclature: IUPAC Names • The more complex alkyl group is the parent name • The group with the oxygen becomes an alkoxy group © 2016 Cengage Learning 5 Nomenclature: Cyclic Ethers (Heterocycles) • Heterocyclic: Oxygen is part of the ring. O • Epoxides (oxiranes) H2C CH2 O • Oxetanes • Furans (Oxolanes) O O • Pyrans (Oxanes) O O O • Dioxanes O © 2013 Pearson Education, Inc. 6 Epoxide Nomenclature • Name the starting alkene and add “oxide” © 2013 Pearson Education, Inc. 7 Epoxide Nomenclature • The oxygen can be treated as a substituent (epoxy) on the compound • Use numbers to specify position • Oxygen is 1, the carbons are 2 and 3 • Substituents are named in alphabetical order © 2013 Pearson Education, Inc. 8 Properties of Ethers • Possess nearly the same geometry as water – Oxygen atom is sp3-hybridized – Bond angles of R–O–R bonds are approximately tetrahedral • Polar C—O bonds © 2013 Pearson Education, Inc. 9 Properties of Ethers: Hydrogen Bond • Hydrogen bond is a attractive interaction between an electronegative atom and a hydrogen atom bonded to another electronegative atom • Ethers cannot hydrogen bond with other ether molecules, so they have a lower boiling point than alcohols • Ether molecules can hydrogen bond with water and alcohol molecules • They are hydrogen bond acceptors © 2013 Pearson Education, Inc. -

Chapter 8: Reactions of Alkanes; Radicals Page
REACTIONS OF ALKANES: CONTENTS Reactivity Considerations Chlorination and Bromination of Alkanes Reactivity–Selectivity Principle Radical Substitution of Benzylic and Allylic Hydrogens Stereochemistry of Radical Substitution No Radical Reactions in Biological Systems 2 RADICALS Sources include: Hydrogen peroxide . Alkyl peroxides . Light causes homolysis of the weak O-O bond . 3 HETEROLYSIS & HOMOLYSIS Heterolytic bond cleavage or heterolysis H Br H+ + Br Homolytic bond cleavage or homolysis H Br H + Br Homolysis produces radicals, which are very reactive species 4 RADICAL CHAIN REACTIONS Initiation turns stable species into radicals by breaking a bond. 5 RADICAL CHAIN REACTIONS Propagation causes products to form without resulting in net consumption of radicals. (production = consumption) 6 RADICAL CHAIN REACTIONS Termination results in net radical destruction. Generally occurs when reactants are used up. 7 CHLORINATION AND BROMINATION OF ALKANES C H A P T E R 12 8 PRODUCT DISTRIBUTION 9 CHLORINATION RATES 10 CHLORINATION PRODUCT DISTRIBUTION 11 BROMINATION RATES 12 REACTIVITY–SELECTIVITY PRINCIPLE The very reactive chlorine atom will have lower selectivity and attack pretty much any hydrogen available on an alkane The less reactive bromine atom will be more selective and tends to react preferentially with the easy targets, i.e. benzylic/allylic > 3° > 2° > 1° 13 RADICALS Stability of alkyl radicals is similar to stability of carbocations 14 CRUDE RADICAL STABILITY INDEX Add 1 for each attached carbon. Add 3 for adjacent double bond or phenyl ring. Radical equally stable on double bond carbon as on single bonded carbon Profile similar to carbocations but resonance contributes more to radical stability, and radical is OK on C=C. -

Pph3-Assisted Esterification of Acyl Fluorides with Ethers Via C
Article PPh3‐Assisted Esterification of Acyl Fluorides with Ethers via C(sp3)–O Bond Cleavage Accelerated by TBAT Zhenhua Wang 1, Xiu Wang 1 and Yasushi Nishihara 2,* 1 Graduate School of Natural Science and Technology, Okayama University, 3‐1‐1 Tsushimanaka, Kita‐ku, Okayama 700‐8530, Japan; [email protected]‐u.ac.jp (Z.W.); [email protected]‐u.ac.jp (X.W.) 2 Research Institute for Interdisciplinary Science, Okayama University, 3‐1‐1 Tsushimanaka, Kita‐ku, Okayama 700‐8530, Japan * Correspondence: ynishiha@okayama‐u.ac.jp; Tel.: +81‐86‐251‐7855; Fax: +81‐86‐251‐7855 Received: 23 May 2019; Accepted: 26 June 2019; Published: 28 June 2019 Abstract: We describe the (triphenylphosphine (PPh3)‐assisted methoxylation of acyl fluorides with cyclopentyl methyl ether (CPME) accelerated by tetrabutylammonium difluorotriphenysilicate (TBAT) via regiospecific C–OMe bond cleavage. Easily available CPME is utilized not only as the solvent, but a methoxylating agent in this transformation. The present method is featured by C–O and C–F bond cleavage under metal‐free conditions, good functional‐group tolerance, and wide substrate scope. Mechanistic studies revealed that the radical process was not involved. Keywords: Acyl fluorides; cyclopentyl methyl ether (CPME); tetrabutylammonium difluorotriphenysilicate (TBAT); carbon‐oxygen bond cleavage; esterification 1. Introduction The C−O bond cleavage in ethers is one of the most fundamental transformations in organic synthesis and has been widely applied in the manufacturing of fine chemicals as well as the synthesis of polyfunctional molecules [1–5]. Particularly, the preparation and degradation of ethers have often been considered important synthetic strategies for the protection/deprotection of hydroxyl groups. -
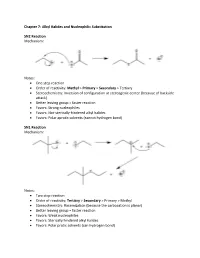
Alkyl Halides and Nucleophilic Substitution SN2 Reaction
Chapter 7: Alkyl Halides and Nucleophilic Substitution SN2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Methyl > Primary > Secondary > Tertiary • Stereochemistry: Inversion of configuration at stereogenic center (because of backside attack) • Better leaving group = faster reaction • Favors: Strong nucleophiles • Favors: Not-sterically-hindered alkyl halides • Favors: Polar aprotic solvents (cannot hydrogen bond) SN1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary > Methyl • Stereochemistry: Racemization (because the carbocation is planar) • Better leaving group = faster reaction • Favors: Weak nucleophiles • Favors: Sterically hindered alkyl halides • Favors: Polar protic solvents (can hydrogen bond) Important Trends Chapter 8: Alkyl Halides and Elimination Reactions E2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: antiperiplanar arrangement of H and X • Better leaving group = faster reaction • Favors: Polar aprotic solvents, strong bases • Products follow Zaitsev rule (more substituted alkene is the major product) E1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: Trigonal planar carbocation intermediate • Better leaving group = faster reaction • Favors: Polar protic solvents, weak bases • Products follow Zaitsev rule Chapter 9: Alcohols, Ethers, and Epoxides Preparation of Alcohols Mechanism: Notes: • SN2 mechanism -

SYNTHESIS, SPECTROSCOPIC INVESTIGATION and IMMOBILIZATION of COPPER(II) COMPLEXES AS OXIDATION CATALYSTS Except Where Reference
SYNTHESIS, SPECTROSCOPIC INVESTIGATION AND IMMOBILIZATION OF COPPER(II) COMPLEXES AS OXIDATION CATALYSTS Except where reference is made to the work of others, the work described in this dissertation is my own or was done in collaboration with my advisory committee. This dissertation does not include proprietary or classified information. ⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯ Moses G. Gichinga Certificate of approval: ⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯ ⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯ Peter Livant Susanne Striegler, Chair Associate Professor Assistant Professor Chemistry and Biochemistry Chemistry and Biochemistry ⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯ ⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯ David Stanbury S. Davis Worley Professor Professor Chemistry and Biochemistry Chemistry and Biochemistry ⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯ George Flowers Dean Graduate School SYNTHESIS, SPECTROSCOPIC INVESTIGATION AND IMMOBILIZATION OF COPPER(II) COMPLEXES AS OXIDATION CATALYSTS Moses G. Gichinga A Dissertation Submitted to the Graduate Faculty of Auburn University in Partial Fulfillments of the Requirements for the Degree of Doctor of Philosophy Auburn, AL August 10, 2009 SYNTHESIS, SPECTROSCOPIC INVESTIGATION AND IMMOBILIZATION OF COPPER(II) COMPLEXES AS OXIDATION CATALYSTS Moses G. Gichinga Permission is granted to Auburn University to make copies of this dissertation at its discretion, upon request of individuals or institutions at their expense. The author reserves all publication rights. ⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯ Signature of Author ⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯ Date of Graduation iii VITA Moses Gichinga, son of Joseph Gichinga and Hannah Wangui, was born on June 2, 1980 in Kenya. He graduated with a Bachelor of Science degree in Chemistry in May, 2004 from University of Nairobi, Kenya. In the fall of 2004, he entered the Graduate School in the Department of Chemistry and Biochemistry at Auburn University. In January of 2005, he joined the laboratory of Dr. Susanne Striegler and is currently pursuing a Ph.D. degree. iv DISSERTATION ABSTRACT SYNTHESIS, SPECTROSCOPIC INVESTIGATION AND IMMOBILIZATION OF COPPER(II) COMPLEXES AS OXIDATION CATALYSTS Moses G.