A Pathogenic Dyskerin Mutation Impairs Proliferation and Activates a DNA Damage Response Independent of Telomere Length in Mice
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

How Shelterin Protects Mammalian Telomeres 303 ANRV361-GE42-15 ARI 3 October 2008 10:10 (See 3' 5' TRF2 S Mplex
ANRV361-GE42-15 ARI 3 October 2008 10:10 ANNUAL How Shelterin Protects REVIEWS Further Click here for quick links to Annual Reviews content online, Mammalian Telomeres including: • Other articles in this volume 1 • Top cited articles Wilhelm Palm and Titia de Lange • Top downloaded articles • Our comprehensive search Laboratory for Cell Biology and Genetics, The Rockefeller University, New York, NY 10021; email: [email protected] 1Current address: Max Planck Institute of Molecular Cell Biology and Genetics, 01307 Dresden, Germany Annu. Rev. Genet. 2008. 42:301–34 Key Words First published online as a Review in Advance on ATM, ATR, cancer, NHEJ, HR August 4, 2008 by Rockefeller University on 10/13/09. For personal use only. The Annual Review of Genetics is online at Abstract genet.annualreviews.org The genomes of prokaryotes and eukaryotic organelles are usually cir- This article’s doi: cular as are most plasmids and viral genomes. In contrast, the nuclear Annu. Rev. Genet. 2008.42:301-334. Downloaded from arjournals.annualreviews.org 10.1146/annurev.genet.41.110306.130350 genomes of eukaryotes are organized on linear chromosomes, which Copyright c 2008 by Annual Reviews. require mechanisms to protect and replicate DNA ends. Eukaryotes All rights reserved navigate these problemswith the advent of telomeres, protective nucle- 0066-4197/08/1201-0301$20.00 oprotein complexes at the ends of linear chromosomes, and telomerase, the enzyme that maintains the DNA in these structures. Mammalian telomeres contain a specific protein complex, shelterin, that functions to protect chromosome ends from all aspects of the DNA damage re- sponse and regulates telomere maintenance by telomerase. -

Dyskerin Mutations Present in Dyskeratosis Congenita Patients Increase Oxidative Stress and DNA Damage Signalling in Dictyostelium Discoideum
cells Article Dyskerin Mutations Present in Dyskeratosis Congenita Patients Increase Oxidative Stress and DNA Damage Signalling in Dictyostelium Discoideum Javier Rodriguez-Centeno, Rosario Perona and Leandro Sastre * Instituto de Investigaciones Biomedicas, CSIC/UAM and Centro de Investigación Biomédica en Red de Enfermedades Raras, CIBERER, 28029 Madrid, Spain; [email protected] (J.R.-C.); [email protected] (R.P.) * Correspondence: [email protected]; Tel.: +3491-585-4437 Received: 11 October 2019; Accepted: 5 November 2019; Published: 8 November 2019 Abstract: Dyskerin is a protein involved in the formation of small nucleolar and small Cajal body ribonucleoproteins. These complexes participate in RNA pseudouridylation and are also components of the telomerase complex required for telomere elongation. Dyskerin mutations cause a rare disease, X-linked dyskeratosis congenita, with no curative treatment. The social amoeba Dictyostelium discoideum contains a gene coding for a dyskerin homologous protein. In this article D. discoideum mutant strains that have mutations corresponding to mutations found in dyskeratosis congenita patients are described. The phenotype of the mutant strains has been studied and no alterations were observed in pseudouridylation activity and telomere structure. Mutant strains showed increased proliferation on liquid culture but reduced growth feeding on bacteria. The results obtained indicated the existence of increased DNA damage response and reactive oxygen species, as also reported in human Dyskeratosis congenita cells and some other disease models. These data, together with the haploid character of D. discoideum vegetative cells, that resemble the genomic structure of the human dyskerin gene, located in the X chromosome, support the conclusion that D. discoideum can be a good model system for the study of this disease. -

Single-Molecule Analysis of the Human Telomerase RNA Center Dot Dyskerin Interaction and the Effect of Dyskeratosis Congenita Mu
Single-Molecule Analysis of the Human Telomerase RNA center dot Dyskerin Interaction and the Effect of Dyskeratosis Congenita Mutations Ashbridge, B; Orte, A; Yeoman, JA; Kirwan, M; Vulliamy, T; Dokal, I; Klenerman, D; Balasubramanian, S © 2009 American Chemical Society http://pubs.acs.org/page/policy/authorchoice_termsofuse.html For additional information about this publication click this link. http://qmro.qmul.ac.uk/xmlui/handle/123456789/14851 Information about this research object was correct at the time of download; we occasionally make corrections to records, please therefore check the published record when citing. For more information contact [email protected] 10858 Biochemistry 2009, 48, 10858–10865 DOI: 10.1021/bi901373e Single-Molecule Analysis of the Human Telomerase RNA 3 Dyskerin Interaction and the Effect of Dyskeratosis Congenita Mutations† Beth Ashbridge,‡,^ Angel Orte,‡,^,@ Justin A. Yeoman,‡,^,# Michael Kirwan, ) Tom Vulliamy, ) Inderjeet Dokal, ) David Klenerman,*,‡,r and Shankar Balasubramanian*,‡,§,r ‡University Chemical Laboratories, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, U.K., §School of Clinical Medicine, University of Cambridge, Cambridge CB2 0SP, U.K., and Centre) for Paediatrics, Institute of Cell and Molecular Science, Barts and The London School of Medicine and Dentistry, Queen Mary, University of London, London E1 2AT, U.K. ^These authors contributed equally to this work. @Present address: Department of Physical Chemistry, Faculty of Pharmacy, Campus Cartuja, Granada 18071, Spain. #Present address: National Centre for Biological Sciences, Tata Institute of Fundamental Research, UAS GKVK, Bellary Road, Bangalore 560 065, India. rJoint senior authorship. Received August 7, 2009; Revised Manuscript Received October 1, 2009 ABSTRACT: It has been proposed that human telomerase RNA (hTR) interacts with dyskerin, prior to assembly of the telomerase holoenzyme. -

Transcriptome-Wide Profiling of Multiple RNA Modifications Simultaneously at Single-Base Resolution,” by Vahid Khoddami, Archana Yerra, Timothy L
Correction BIOCHEMISTRY, CHEMISTRY Correction for “Transcriptome-wide profiling of multiple RNA modifications simultaneously at single-base resolution,” by Vahid Khoddami, Archana Yerra, Timothy L. Mosbruger, Aaron M. Fleming, Cynthia J. Burrows, and Bradley R. Cairns, which was first published March 14, 2019; 10.1073/pnas.1817334116 (Proc Natl Acad Sci USA 116:6784–6789). The authors note that the following statement should be added to the Acknowledgments: “This work was also supported by Grant R01 GM093099 from the NIH/National Institute of General Medical Sciences (to C.J.B.).” Published under the PNAS license. Published online April 22, 2019. www.pnas.org/cgi/doi/10.1073/pnas.1905628116 9136 | PNAS | April 30, 2019 | vol. 116 | no. 18 www.pnas.org Downloaded by guest on October 2, 2021 Transcriptome-wide profiling of multiple RNA modifications simultaneously at single-base resolution Vahid Khoddamia,b,c,1,2, Archana Yerrab,c,1, Timothy L. Mosbrugerd, Aaron M. Fleminge, Cynthia J. Burrowse,3, and Bradley R. Cairnsb,c,3 aDepartment of Cell Biology, Harvard Medical School, Boston, MA 02115; bHoward Hughes Medical Institute, University of Utah School of Medicine, Salt Lake City, UT 84112; cDepartment of Oncological Sciences, Huntsman Cancer Institute, University of Utah School of Medicine, Salt Lake City, UT 84112; dBioinformatics Shared Resource, Huntsman Cancer Institute, University of Utah School of Medicine, Salt Lake City, UT 84112; and eDepartment of Chemistry, University of Utah, Salt Lake City, UT 84112 Contributed by Cynthia J. Burrows, January 25, 2019 (sent for review October 9, 2018; reviewed by Juan D. Alfonzo, Thomas Carell, and Peter C. -

Qt38n028mr Nosplash A3e1d84
! ""! ACKNOWLEDGEMENTS I dedicate this thesis to my parents who inspired me to become a scientist through invigorating scientific discussions at the dinner table even when I was too young to understand what the hippocampus was. They also prepared me for the ups and downs of science and supported me through all of these experiences. I would like to thank my advisor Dr. Elizabeth Blackburn and my thesis committee members Dr. Eric Verdin, and Dr. Emmanuelle Passegue. Liz created a nurturing and supportive environment for me to explore my own ideas, while at the same time teaching me how to love science, test my questions, and of course provide endless ways to think about telomeres and telomerase. Eric and Emmanuelle both gave specific critical advice about the proper experiments for T cells and both volunteered their lab members for further critical advice. I always felt inspired with a sense of direction after thesis committee meetings. The Blackburn lab is full of smart and dedicated scientists whom I am thankful for their support. Specifically Dr. Shang Li and Dr. Brad Stohr for their stimulating scientific debates and “arguments.” Dr. Jue Lin, Dana Smith, Kyle Lapham, Dr. Tet Matsuguchi, and Kyle Jay for their friendships and discussions about what my data could possibly mean. Dr. Eva Samal for teaching me molecular biology techniques and putting up with my late night lab exercises. Beth Cimini for her expertise with microscopy, FACs, singing, and most of all for being a caring and supportive friend. Finally, I would like to thank Dr. Imke Listerman, my scientific partner for most of the breast cancer experiments. -

Mutations in the Telomerase Component NHP2 Cause the Premature Ageing Syndrome Dyskeratosis Congenita
Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita Tom Vulliamy†‡, Richard Beswick†, Michael Kirwan†, Anna Marrone†§, Martin Digweed¶, Amanda Walne†, and Inderjeet Dokal† †Academic Unit of Paediatrics, Institute of Cell and Molecular Science, Barts and the London School of Medicine and Dentistry, London E1 2AT, United Kingdom; and ¶Institut fu¨r Humangenetik, Charite´-Universita¨tsmedizin Berlin, Campus Virchow-Klinikum, 13353 Berlin, Germany Edited by Ernest Beutler, The Scripps Research Institute, La Jolla, CA, and approved April 14, 2008 (received for review January 3, 2008) Dyskeratosis congenita is a premature aging syndrome character- congenita patients have very short telomeres (10, 15), and some ized by muco-cutaneous features and a range of other abnormal- have been shown to have reduced levels of TERC (the RNA ities, including early greying, dental loss, osteoporosis, and malig- component of telomerase) (16, 17). It has been suggested nancy. Dyskeratosis congenita cells age prematurely and have very therefore that dyskeratosis congenita may primarily be a disor- short telomeres. Patients have mutations in genes that encode der of telomere maintenance. The most compelling evidence components of the telomerase complex (dyskerin, TERC, TERT, and supporting this view is that autosomal dominant dyskeratosis NOP10), important in the maintenance of telomeres. Many dys- congenita can result from TERC mutations (18), which cause a keratosis congenita patients remain uncharacterized. Here, we reduction in telomerase activity and give rise to disease via describe the analysis of two other proteins, NHP2 and GAR1, that haploinsufficiency (19–21). Heterozygous missense mutations in together with dyskerin and NOP10 are key components of telom- the reverse transcriptase component of telomerase (TERT) that erase and small nucleolar ribonucleoprotein (snoRNP) complexes. -

1 Novel Expression Signatures Identified by Transcriptional Analysis
ARD Online First, published on October 7, 2009 as 10.1136/ard.2009.108043 Ann Rheum Dis: first published as 10.1136/ard.2009.108043 on 7 October 2009. Downloaded from Novel expression signatures identified by transcriptional analysis of separated leukocyte subsets in SLE and vasculitis 1Paul A Lyons, 1Eoin F McKinney, 1Tim F Rayner, 1Alexander Hatton, 1Hayley B Woffendin, 1Maria Koukoulaki, 2Thomas C Freeman, 1David RW Jayne, 1Afzal N Chaudhry, and 1Kenneth GC Smith. 1Cambridge Institute for Medical Research and Department of Medicine, Addenbrooke’s Hospital, Hills Road, Cambridge, CB2 0XY, UK 2Roslin Institute, University of Edinburgh, Roslin, Midlothian, EH25 9PS, UK Correspondence should be addressed to Dr Paul Lyons or Prof Kenneth Smith, Department of Medicine, Cambridge Institute for Medical Research, Addenbrooke’s Hospital, Hills Road, Cambridge, CB2 0XY, UK. Telephone: +44 1223 762642, Fax: +44 1223 762640, E-mail: [email protected] or [email protected] Key words: Gene expression, autoimmune disease, SLE, vasculitis Word count: 2,906 The Corresponding Author has the right to grant on behalf of all authors and does grant on behalf of all authors, an exclusive licence (or non-exclusive for government employees) on a worldwide basis to the BMJ Publishing Group Ltd and its Licensees to permit this article (if accepted) to be published in Annals of the Rheumatic Diseases and any other BMJPGL products to exploit all subsidiary rights, as set out in their licence (http://ard.bmj.com/ifora/licence.pdf). http://ard.bmj.com/ on September 29, 2021 by guest. Protected copyright. 1 Copyright Article author (or their employer) 2009. -

Telomere Maintenance Pathway Activity Analysis Enables Tissue- and Gene-Level Inferences
bioRxiv preprint doi: https://doi.org/10.1101/2021.02.01.429081; this version posted February 2, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Telomere maintenance pathway activity analysis enables tissue- and gene-level inferences Lilit Nersisyan1,2*, Arman Simonyan1, Hans Binder3, Arsen Arakelyan1,2 1 Bioinformatics Group, Institute of Molecular Biology, National Academy of Sciences, Yerevan, Armenia 2 Pathverse, LLC, Yerevan, Armenia 3 Interdisciplinary Center for Bioinformatics, University of Leipzig, Leipzig, Germany * Correspondence: Lilit Nersisyan Keywords: telomere maintenance mechanisms, telomerase, alternative lengthening of telomeres, pathway signal flow, testis ABSTRACT Telomere maintenance is one of the mechanisms ensuring indefinite divisions of cancer and stem cells. Good understanding of telomere maintenance mechanisms (TMM) is important for studying cancers and designing therapies. However, molecular factors triggering selective activation of either the telomerase dependent (TEL) or the alternative lengthening of telomeres (ALT) pathway are poorly understood. In addition, more accurate and easy-to-use methodologies are required for TMM phenotyping. In this study, we have performed literature based reconstruction of signaling pathways for the ALT and TEL TMMs. Gene expression data were used for computational assessment of TMM pathway activities and compared with experimental assays for TEL and ALT. Explicit consideration of pathway topology makes bioinformatics analysis more informative compared to computational methods based on simple summary measures of gene expression. Application to healthy human tissues showed high ALT and TEL pathway activities in testis, and identified genes and pathways that may trigger TMM activation. -
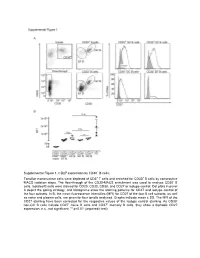
Supplemental Figure 1. CD27 Expression by CD30+ B Cells
Supplemental Figure 1. CD27 expression by CD30+ B cells. Tonsillar mononuclear cells were depleted of CD3+ T cells and enriched for CD30+ B cells by consecutive MACS isolation steps. The flow-through of the CD30-MACS enrichment was used to analyze CD30- B cells. Isolated B cells were stained for CD20, CD30, CD38, and CD27 or isotype control. Dot plots in panel A depict the gating strategy, and histograms show the staining patterns for CD27 and isotype control of the four subsets. In B, the mean fluorescence intensities (MFI) for CD27 of the four B cell subsets, as well as naive and plasma cells, are given for four tonsils analyzed. Graphs indicate mean ± SD. The MFI of the CD27 staining have been corrected for the respective values of the isotype control staining. As CD30- non-GC B cells include CD27- naïve B cells and CD27+ memory B cells, they show a biphasic CD27 expression. n.s., not significant; ** p<0.01 (unpaired t test). Supplemental Figure 2. Interleukin receptor expression by CD30+ B cells and relatedness of CD30+ EF B cells to CD21low B cells. mRNA and surface protein expression of IL2RB (A), IL21R (B) and CD21 (C) are shown for indicated B cell subsets of five tonsils (conventional (conv.) CD30- GC (germinal center) B cells (CD20highCD38+), CD30+ GC B cells, CD30- memory and CD30+ EF (extra follicular) B cells (CD20+CD38-/lowCD27+), and plasma cells (PC) (CD20+CD38high). mRNA data originate from Affymetrix genechip analyses (Tiacci et al., Blood, 2012; 120:4609-4620) and were analyzed using an unpaired t test. -

DKC1 Gene Dyskerin Pseudouridine Synthase 1
DKC1 gene dyskerin pseudouridine synthase 1 Normal Function The DKC1 gene provides instructions for making a protein called dyskerin. This protein is involved in maintaining structures called telomeres, which are found at the ends of chromosomes. Telomeres help protect chromosomes from abnormally sticking together or breaking down (degrading). In most cells, telomeres become progressively shorter as the cell divides. After a certain number of cell divisions, the telomeres become so short that they trigger the cell to stop dividing or to self-destruct (undergo apoptosis). Telomeres are maintained by two important protein complexes, telomerase and shelterin. Telomerase counteracts the shortening of telomeres by adding small repeated segments of DNA to the ends of chromosomes each time the cell divides. One component of telomerase, called hTR, provides a template for creating the repeated sequence of DNA that telomerase adds to the ends of chromosomes. The dyskerin protein attaches (binds) to hTR and helps stabilize the telomerase complex. In most types of cells, telomerase is either undetectable or active at very low levels. However, telomerase is highly active in cells that divide rapidly, such as cells that line the lungs and gastrointestinal tract, cells in bone marrow, and cells of the developing fetus. Telomerase allows these cells to divide many times without becoming damaged or undergoing apoptosis. Telomerase is also abnormally active in most cancer cells, which grow and divide without control or order. Dyskerin is also involved in the production of ribosomal RNA (rRNA), a chemical cousin of DNA. Ribosomal RNA is required for assembling protein building blocks (amino acids) into functioning proteins. -

Molecular Basis of Telomere Dysfunction in Human Genetic Diseases
FOCUS ON TELOMERES RE V IE W Molecular basis of telomere dysfunction in human genetic diseases Grzegorz Sarek1,2, Paulina Marzec1,2, Pol Margalef1,2 & Simon J Boulton1 Mutations in genes encoding proteins required for telomere structure, replication, repair and length maintenance are associated with several debilitating human genetic disorders. These complex telomere biology disorders (TBDs) give rise to critically short telomeres that affect the homeostasis of multiple organs. Furthermore, genome instability is often a hallmark of telomere syndromes, which are associated with increased cancer risk. Here, we summarize the molecular causes and cellular consequences of disease-causing mutations associated with telomere dysfunction. In contrast to the circular genomes of prokaryotes, eukaryotic is reexpressed, thus allowing maintenance of telomere length and genomes are organized into linear chromosomes, which require mech- enabling cancer-cell immortalization and disease progression13,14. anisms to protect and maintain the chromosome ends. Eukaryotes have solved this problem with telomeres, complex nucleoprotein Shelterin structures that protect chromosome ends from promiscuous DNA- Telomere function and integrity are critically dependent on a repair activities and nucleolytic degradation (reviewed in ref. 1). six-subunit protein complex called shelterin, which specifically Telomeres are formed by tandem-repeat sequences (TTAGGG in associates with telomeric repeats (Fig. 2). Shelterin comprises vertebrates) and terminate in a 3′ single-stranded DNA overhang on telomere repeat–binding factor (TRF) 1 (TRF1), TRF2, protection of the G-rich strand2,3. A major challenge to chromosome integrity is telomeres 1 (POT1), TRF1-interacting nuclear factor 2 (TIN2), repressor the progressive shortening of telomeres with each cell division; this activator protein 1 (Rap1) and TINF2-interacting protein 2 (TPP1) shortening eventually triggers cellular senescence4. -

ROLE of Hnrnp A/B PROTEINS in TELOMERE MAINTENANCE and TELOMERASE ACTIVITY
ROLE OF hnRNP A/B PROTEINS IN TELOMERE MAINTENANCE AND TELOMERASE ACTIVITY by EMAD AHMED SAIG (BSc and MSc) A thesis submitted for the degree of Master of Philosophy at The University of Queensland (2018) School of Chemistry and Molecular Bioscience Abstract Telomere shortening leads to eukaryotic cell senescence, whereas enhanced telomerase activity is associated with telomere expansion and growth of cancer cells. Several studies have suggested hnRNPs are important for telomere maintenance including their ability to bind telomeres, and telomerase. The hnRNPs A/B family are highly abundant multifunctional proteins that bind to single-stranded DNA and RNA and perform many roles in cellular regulation. In this study, affinity pull-down assays, biosensor and UV-cross-linking studies were used to confirm that recombinant hnRNPs A2 and A3, compared to recombinant hnRNP A1, specifically interact with single-stranded telomeric DNA repeat TTAGGG and hexamer repeat. Tandem RRMs are required for strong binding, which is little changed by exclusion of the glycine-rich domain (hnRNPs Δ GRD). Additionally, TRAP assays, that indicate telomerase activity, showed that hnRNPs A1, A2 and A3 bind to telomerase in cell extracts. It is not clear whether this association is mediated by binding a specific nucleotide region within telomerase RNA, or a protein component such as dyskerin, or both. In this study I have confirmed and refined the interaction site between the hnRNP A/B proteins and telomerase RNA (hTR). Serial UV-cross-linking RNA electrophoretic mobility shifting assays (REMSA) have shown that hnRNP A2 binds specifically with to a 17nt region within the RNA telomerase template (hTR).