New Synthetic Technology for the Synthesis of Hindered R
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Methanesulfonyl Chloride
A Publication of Reliable Methods for the Preparation of Organic Compounds Working with Hazardous Chemicals The procedures in Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices. In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red “Caution Notes” within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices. The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein. -

덴드리틱 벤질 클로라이드의 효율적인 합성 Facile Synthesis of Dendritic Benzyl Chlorides from Their Alcohols W
Polymer(Korea), Vol. 31, No. 5, pp 417-421, 2007 덴드리틱 벤질 클로라이드의 효율적인 합성 이재욱F,7ㆍ한승철Fㆍ김희주ㆍ김정환ㆍ이언엽ㆍ김병기ㆍ성새름ㆍ강화신ㆍ김지현FFㆍ허도성FFF 동아대학교 화학과, F동아대학교 의생명과학과, FF동국대학교 생명화학공학과, FFF인제대학교 의생명화학과 (2007년 5월 9일 접수, 2007년 6월 29일 채택) Facile Synthesis of Dendritic Benzyl Chlorides from Their Alcohols with Methanesulfonyl Chloride/Et3N Jae Wook Lee*,7, Seung Choul Han*, Hee Joo Kim, Jung Hwan Kim, Un Yup Lee, Byoung-Ki Kim, Sae Reum Sung, Hwa-Shin Kang, Ji Hyeon Kim**, and Do-Sung Huh*** Department of Chemistry, Dong-A University, Hadan-2-dong, Busan 604-714, Korea *Department of Medical Bioscience, Dong-A University, Hadan-2-dong, Busan 604-714, Korea **Department of Chemical & Biochemical Engineering, Dongguk University, Seoul 100-715, Korea ***Department of Biomedicinal Chemistry, Inje University, Gim Hai 621-749, Korea (Received May 9, 2007; Accepted June 29, 2007) 초록: 덴드리틱 벤질 알코올을 트리에틸아민과 메탄술포닐클로라이드와 반응시켜서, 덴드리틱 벤질 클로라이드 의 효율적인 합성이 이루어졌다. 이 반응은 히드록시기의 메실화 반응과 염소화 반응의 2단계 반응으로 이루어지 는데, 중간체의 분리없이 한 반응 용기내에서 반응이 진행되는 경제적인 방법이다. Abstract: A successful rapid synthesis of dendritic benzyl chlorides from dendritic benzyl alcohols using methanesulfonyl chloride/Et3N as activating agents was described. In this method, each dendritic benzyl chloride can be prepared in one pot: no isolation of intermediate mesylated dendrons is required. The key steps in the syntheses of dendritic benzyl chlorides were the mesylation of the hydroxymethyl group followed by the chlorination by in-situ generated triethylammonium chloride. Keywords: benzylic alcohol, chlorination. Introduction vation and coupling steps in high yield. In addition, the coupling step must be very efficient to enable complete Dendrimers represent a novel type of polymeric material reaction. -
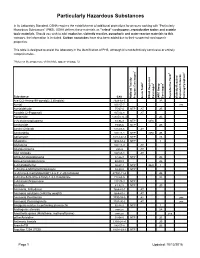
Particularly Hazardous Substances
Particularly Hazardous Substances In its Laboratory Standard, OSHA requires the establishment of additional protections for persons working with "Particularly Hazardous Substances" (PHS). OSHA defines these materials as "select" carcinogens, reproductive toxins and acutely toxic materials. Should you wish to add: explosive, violently reactive, pyrophoric and water-reactve materials to this category, the information is included. Carbon nanotubes have also been added due to their suspected carcinogenic properties. This table is designed to assist the laboratory in the identification of PHS, although it is not definitively conclusive or entirely comprehensive. *Notes on the proper use of this table appear on page 12. 1 6 5 2 3 4 Substance CAS National Toxicity National Program Carcinogen Toxin Acute Regulated OSHA Carcinogen Group IARC Carcinogen Toxin Reproductive Violently Reactive/ Explosive/Peroxide Forming/Pyrophoric A-a-C(2-Amino-9H-pyrido[2,3,b]indole) 2648-68-5 2B Acetal 105-57-7 yes Acetaldehyde 75-07-0 NTP AT 2B Acrolein (2-Propenal) 107-02-8 AT Acetamide 126850-14-4 2B 2-Acetylaminofluorene 53-96-3 NTP ORC Acrylamide 79-06-6 NTP 2B Acrylyl Chloride 814-68-6 AT Acrylonitrile 107-13-1 NTP ORC 2B Adriamycin 23214-92-8 NTP 2A Aflatoxins 1402-68-2 NTP 1 Allylamine 107-11-9 AT Alkylaluminums varies AT Allyl Chloride 107-05-1 AT ortho-Aminoazotoluene 97-56-3 NTP 2B para-aminoazobenzene 60-09-3 2B 4-Aminobiphenyl 92-67-1 NTP ORC 1 1-Amino-2-Methylanthraquinone 82-28-0 NTP (2-Amino-6-methyldipyrido[1,2-a:3’,2’-d]imidazole) 67730-11-4 2B -

Fragment-Based Discovery of a New Class of Inhibitors Targeting Mycobacterial Trna Modification
Supplementary Data Fragment-based discovery of a new class of inhibitors targeting mycobacterial tRNA modification Sherine E. Thomas*1, Andrew J. Whitehouse*2, Karen Brown3,4, Juan M. Belardinelli5, Ramanuj Lahiri6, M. Daben J. Libardo7, Pooja Gupta1, Sony Malhotra8, Helena I. M. Boshoff7, Mary Jackson5, Chris Abell 2, Anthony G. Coyne2, Tom L. Blundell §1, R. Andres Floto3,4, Vitor Mendes §1 1 Department of Biochemistry, University of Cambridge, 80 Tennis Court Road, Cambridge CB2 1GA, UK. 2 Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. 3 MRC Laboratory of Molecular Biology, Francis Crick Avenue, Cambridge, CB2 0QH, U.K. 4 Cambridge Centre for Lung Infection, Royal Papworth Hospital, Cambridge, CB23 3RE, U.K. 5 Mycobacteria Research Laboratories, Department of Microbiology, Immunology and Pathology, Colorado State University, Fort Collins, Colorado, USA. 6 National Hansen’s Disease Program, Healthcare Systems Bureau, HRSA, DHHS, USA 7 Tuberculosis Research Section, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Disease, National Institutes of Health, 9000 Rockville Pike, Bethesda, Maryland 20892, USA. 8 Birkbeck College, University of London, Malet Street, WC1E7HX, UK * Contributed equally § To whom correspondence should be addressed Results Conservation of catalytic residues in mycobacterial TrmD A multiple sequence alignment of 5 mycobacterial TrmD enzyme sequences with 10 TrmD ortholog amino acid sequences (Figure S1) shows conservation of important catalytic residues and residues involved in tRNA recognition and methyl transfer reactions. The catalytic residues Asp169 and Arg154 are highly conserved across TrmD orthologs. Key residues such as Asp50, Gly59, His46 involved in tRNA G36 and G37 base recognition, interactions with bases at positions 38, 32 and anticodon branch of wild type tRNA respectively, are also broadly conserved. -

4 Methanesulfonyl Chloride1 Acute Exposure Guideline Levels
Committee on Acute Exposure Guideline Levels Committee on Toxicology Board on Environmental Studies and Toxicology Division on Earth and Life Studies THE NATIONAL ACADEMIES PRESS 500 FIFTH STREET, NW WASHINGTON, DC 20001 NOTICE: The project that is the subject of this report was approved by the Governing Board of the National Research Council, whose members are drawn from the councils of the National Academy of Sciences, the National Academy of Engineering, and the Insti- tute of Medicine. The members of the committee responsible for the report were chosen for their special competences and with regard for appropriate balance. This project was supported by Contract No. W81K04-11-D-0017 and EP-W-09-007 be- tween the National Academy of Sciences and the U.S. Department of Defense and the U.S. Environmental Protection Agency. Any opinions, findings, conclusions, or recom- mendations expressed in this publication are those of the author(s) and do not necessarily reflect the view of the organizations or agencies that provided support for this project. International Standard Book Number-13: 978-0-309-28308-3 International Standard Book Number-10: 0-309-28308-6 Additional copies of this report are available for sale from the National Academies Press, 500 Fifth Street, NW, Keck 360, Washington, DC 20001; (800) 624-6242 or (202) 334- 3313; http://www.nap.edu/. Copyright 2013 by the National Academy of Sciences. All rights reserved. Printed in the United States of America The National Academy of Sciences is a private, nonprofit, self-perpetuating society of distinguished scholars engaged in scientific and engineering research, dedicated to the furtherance of science and technology and to their use for the general welfare. -

Guide for the Selection of Chemical and Biological Decontamination Equipment for Emergency First Responders
U.S. Department of Justice Office of Justice Programs National Institute of Justice National Institute of Justice Law Enforcement and Corrections Standards and Testing Program Guide for the Selection of Chemical and Biological Decontamination Equipment for Emergency First Responders NIJ Guide 103–00 Volume I October 2001 ABOUT THE LAW ENFORCEMENT AND CORRECTIONS STANDARDS AND TESTING PROGRAM The Law Enforcement and Corrections Standards and Testing Program is sponsored by the Office of Science and Technology of the National Institute of Justice (NIJ), U.S. Department of Justice. The program responds to the mandate of the Justice System Improvement Act of 1979, which directed NIJ to encourage research and development to improve the criminal justice system and to disseminate the results to Federal, State, and local agencies. The Law Enforcement and Corrections Standards and Testing Program is an applied research effort that determines the technological needs of justice system agencies, sets minimum performance standards for specific devices, tests commercially available equipment against those standards, and disseminates the standards and the test results to criminal justice agencies nationally and internationally. The program operates through: The Law Enforcement and Corrections Technology Advisory Council (LECTAC), consisting of nationally recognized criminal justice practitioners from Federal, State, and local agencies, which assesses technological needs and sets priorities for research programs and items to be evaluated and tested. The Office of Law Enforcement Standards (OLES) at the National Institute of Standards and Technology, which develops voluntary national performance standards for compliance testing to ensure that individual items of equipment are suitable for use by criminal justice agencies. -

Guide for the Selection of Personal Protective Equipment for Emergency First Responders
U.S. Department of Justice Office of Justice Programs National Institute of Justice National Institute of Justice Law Enforcement and Corrections Standards and Testing Program Guide for the Selection of Personal Protective Equipment for Emergency First Responders NIJ Guide 102–00 Volume I November 2002 U.S. Department of Justice Office of Justice Programs 810 Seventh Street N.W. Washington, DC 20531 John Ashcroft Attorney General Deborah J. Daniels Assistant Attorney General Sarah V. Hart Director, National Institute of Justice For grant and funding information, contact: Department of Justice Response Center 800–421–6770 Office of Justice Programs National Institute of Justice World Wide Web Site World Wide Web Site http://www.ojp.usdoj.gov http://www.ojp.usdoj.gov/nij U.S. Department of Justice Office of Justice Programs National Institute of Justice Guide for the Selection of Personal Protective Equipment for Emergency First Responders NIJ Guide 102-00, Volume I Dr. Alim A. Fatah1 John A. Barrett2 Richard D. Arcilesi, Jr.2 Charlotte H. Lattin2 Charles G. Janney2 Edward A. Blackman2 Coordination by: Office of Law Enforcement Standards National Institute of Standards and Technology Gaithersburg, MD 20899–8102 Prepared for: National Institute of Justice Office of Science and Technology Washington, DC 20531 November 2002 This document was prepared under CBIAC contract number SPO-900-94-D-0002 and Interagency Agreement M92361 between NIST and the Department of Defense Technical Information Center (DTIC). NCJ 191518 1National Institute of Standards and Technology, Office of Law Enforcement Standards. 2Battelle Memorial Institute. National Institute of Justice Sarah V. Hart Director This guide was prepared for the National Institute of Justice, U.S. -

Studies Towards the Synthesis of the Core Structure of Sarains A, B, and C
T v v O T T V\ v r S - j si V V A ^ O Synthetic Studies Toward^he^Core Structure of Sarains A, B and C A Thesis Presented to the University of London in Partial Fulfilment of the Requirements for the Degree of Doctor of Philosophy Gurdeep Singh Nandra September 2004 Christopher Ingold Laboratories Department of Chemistry University College London London W ClH 0AJ SSSSSSSL ProQuest Number: 10014333 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest. ProQuest 10014333 Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC. ProQuest LLC 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106-1346 Abstract Sarains A-C are marine alkaloids produced by the sponge Reniera sarai, which exhibit antitumor, antibacterial, and insecticidal activities. Sarains A-C have a complex structure, which comprises of a l,5-diazatetracyclo[6.2.1.1^’^.0'^’*^]dodecane tricyclic core surrounded by two macrocycles. To date four groups have communicated the construction of the tricyclic core, however the total synthesis of sarains A-C has yet to be reported. This thesis describes the work undertaken towards the synthesis of a tricycle that contains the core structure of sarains A-C, by the use of a novel carbene mediated ylide rearrangement reaction. -

Sustainable Manufacturing at CABB Chlorination and Sulfonation – Surprisingly Sustainable
Sustainable manufacturing at CABB Chlorination and sulfonation – surprisingly sustainable Dr. Jörg Schrickel New Business Development, CABB AG RSC Symposium, Budapest, June 18-19, 2014 Sustainable manufacturing Content Introduction . Sustainability, Green Chemistry and metrics . CABB and CABB‘s Verbund and recycling system Case studies: . Mesylation reaction: batch to continuous . Chlorination reaction I: scrubber to Verbund . Chlorination reaction II: continuous and recycling . Sulfonation reaction: Verbund . Amidation reaction: more or less hazardous Conclusion [ 2 ] Sustainable manufacturing Sustainability Economy, Ecology, Society . Efficient, safe and environmentally benign chemical products and processes . Protecting and enhancing human health and the environment . Reducing the environmental impact of processes and products, minimising waste . Extending the quality of life; competitive, knowledge-based, enterprise-led economy Source: OECD [ 3 ] Sustainable manufacturing 12 Principles of Green Chemistry Prevention of waste: It is better to prevent waste than to treat or clean up waste. Atom Economy: Design of synthesis to maximize the incorporation of all materials used in the process into the final product. Less Hazardous Chemical Syntheses: Design of synthesis to use and generate substances that possess little or no toxicity to human health and the environment. Wherever practicable. Designing Safer Chemicals: Design of chemical products to effect their desired function while minimizing their toxicity. Safer Solvents and Auxiliaries: -

Chemical Compatibility Storage Group
CHEMICAL SEGREGATION Chemicals are to be segregated into 11 different categories depending on the compatibility of that chemical with other chemicals The Storage Groups are as follows: Group A – Compatible Organic Acids Group B – Compatible Pyrophoric & Water Reactive Materials Group C – Compatible Inorganic Bases Group D – Compatible Organic Acids Group E – Compatible Oxidizers including Peroxides Group F– Compatible Inorganic Acids not including Oxidizers or Combustible Group G – Not Intrinsically Reactive or Flammable or Combustible Group J* – Poison Compressed Gases Group K* – Compatible Explosive or other highly Unstable Material Group L – Non-Reactive Flammable and Combustible, including solvents Group X* – Incompatible with ALL other storage groups The following is a list of chemicals and their compatibility storage codes. This is not a complete list of chemicals, but is provided to give examples of each storage group: Storage Group A 94‐75‐7 2,4‐D (2,4‐Dichlorophenoxyacetic acid) 94‐82‐6 2,4‐DB 609-99-4 3,5-Dinitrosalicylic acid 64‐19‐7 Acetic acid (Flammable liquid @ 102°F avoid alcohols, Amines, ox agents see SDS) 631-61-8 Acetic acid, Ammonium salt (Ammonium acetate) 108-24-7 Acetic anhydride (Flammable liquid @102°F avoid alcohols see SDS) 79‐10‐7 Acrylic acid Peroxide Former 65‐85‐0 Benzoic acid 98‐07‐7 Benzotrichloride 98‐88‐4 Benzoyl chloride 107-92-6 Butyric Acid 115‐28‐6 Chlorendic acid 79‐11‐8 Chloroacetic acid 627‐11‐2 Chloroethyl chloroformate 77‐92‐9 Citric acid 5949-29-1 Citric acid monohydrate 57-00-1 Creatine 20624-25-3 -

(12) Patent Application Publication (10) Pub. No.: US 2016/0102064 A1 DODDA Et Al
US 201601 02064A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2016/0102064 A1 DODDA et al. (43) Pub. Date: Apr. 14, 2016 (54) IMPROVED PROCESSES FOR THE Publication Classification PREPARATION OF LINEZOLD USING (51) Int. Cl. NOVELINTERMEDIATES C07D 263/24 (2006.01) C07D 295/135 (2006.01) (71) Applicant: SYMED LABS LIMITED, Hyderabad C07D 209/248 (2006.01) (IN) (52) U.S. Cl. CPC ............ C07D 263/24 (2013.01); C07D 209/48 (72) Inventors: Mohan Rao DODDA, Hyderabad (IN); (2013.01); C07D 295/135 (2013.01) Venkat Reddy BUTHUKURI, (57) ABSTRACT Hyderabad (IN) Provided herein are improved, commercially viable and industrially advantageous processes for the preparation of Linezolid, in high yield and purity, using novel intermediates. (21) Appl. No.: 14/785,960 In one aspect, provided herein are efficient, industrially advantageous and environmentally friendly processes for the (22) PCT Filed: Apr. 25, 2013 preparation of lineZolid, in high yield and with high purity, using novel intermediates. The processes disclosed herein (86). PCT No.: PCT/N2013/000278 avoid the tedious and cumbersome procedures of the prior processes, thereby resolving the problems associated with the S371 (c)(1), processes described in the prior art, which is more convenient (2) Date: Oct. 21, 2015 to operate at lab scale and in commercial scale operations. US 2016/01 02064 A1 Apr. 14, 2016 IMPROVED PROCESSES FOR THE cocci, Vancomycin-resistant enterococci (VRE), and methi PREPARATION OF LINEZOLD USING cillin-resistant Staphylococcus aureus (MRSA). Linezolid is NOVELINTERMEDIATES represented by the following structural formula I: FIELD OF THE INVENTION 0001. The present invention relates to improved, commer cially viable and industrially advantageous processes for the preparation of Linezolid, in high yield and purity, using novel N intermediates. -

A Process for Preparing an Acyl Halide Or Sulfonyl Halide
~™ llll III II II III II III I II II III (19) J European Patent Office Office europeen des brevets (1 1 ) EP0 751 131 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int. CI.6: C07D 233/18, C07C 51/60, 02.01.1997 Bulletin 1997/01 C07C 303/02, C07B 41/1 0 (21) Application number: 96108936.4 (22) Date of filing: 04.06.1996 (84) Designated Contracting States: • Hayashi, Hidetoshi DE FR IT NL Ohmuta-shi, Fukuoka-ken, 836 (JP) • Mizuta, Hideki (30) Priority: 20.06.1995 JP 152826/95 Ohmuta-shi, Fukuoka-ken, 837 (JP) (71) Applicant: MITSUI TOATSU CHEMICALS, Inc. (74) Representative: Strehl Schubel-Hopf Groening & Chiyoda-Ku Tokyo 1 00 (JP) Partner Maximilianstrasse 54 (72) Inventors: 80538 Munchen (DE) • Nagata, Teruyuki Ohmuta-shi, Fukuoka-ken, 836 (JP) (54) A process for preparing an acyl halide or sulfonyl halide (57) A preparation process of acyl halide or sulfonyl halide which comprises reacting corresponding carboxylic acid or sulfonic acid with a haloiminium salt represented by the general formula (1): R'-N -R2 X ( 1 ) XCrU/n wherein R1 and R2 are same or different lower alkyl groups, X is a halogen atom, and n is an integer of 2 or 3. CO LO o Q_ LU Printed by Rank Xerox (UK) Business Services 2.13.10/3.4 EP0 751 131 A1 Description 1 . Field of the Invention 5 The present invention relates to a preparation process of acyl halide or sulfonyl halide. 2. Description of the Related Art In recent years, acyl halide has become important in industry as an intermediate for preparing heat resistant resin, 10 medicines and agricultural chemicals.