Mechanisms of Internal Substitution Reactions Abstract Approved Jor Prof
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

"Alcohol Activation" That Would Be Stereochemically Complementary to That Involving Reaction of an Alcohol with P / S Halides (Notes of Nov 20)
CHEM 203 Topics Discussed on Nov. 23 Desirability of a method for "alcohol activation" that would be stereochemically complementary to that involving reaction of an alcohol with P / S halides (notes of Nov 20): PBr3 CH S Na 3 overall (inversion of H (inversion) H retention configuration) Br SMe (S)-2-bromobutane (R)-configured pdt. H OH (R)-2-butanol ? (S)-configured pdt. overall H inversion SMe Sulfonyl chlorides: para-toluenesulfonyl ("tosyl") chloride, methanesulfonyl ("mesyl") chloride O O O R S Cl H3C S Cl S Cl O O O A generic sulfonyl methanesulfonyl chloride Toluene para-toluenesulfonyl chloride chloride: R = any ( "mesyl chloride" ) (= methyl benzene) ( "tosyl chloride" ) alkyl group Pyridine: a weakly basic, nucleophilic analog of benzene in which an N atom replaces a CH unit: pyridine N Reaction of primary and secondary alcohols with sulfonyl chlorides in the presence of pyridine: formation of sulfonate esters (= alkyl sulfonates): R1 O R1 O OH + Cl S R N O S R + R2 O R2 N O H Cl a generic primary or an "alkyl sulfonate" secondary alcohol ("tosylate", "mesylate," etc.) note: tertiary alcohols are insufficiently nucleophilic to react with sulfonyl chlorides Presumed mechanism for the formation of sulfonate esters from primary and secondary (but not tertiary) alcohols and sulfonyl chlorides: • slow rate of reaction of an alcohol with sulfonyl chlorides in the presence of generic bases Lecture of Nov. 23 p. 2 • pyridine as a nucleophilic catalyst that greatly accelerates the reaction of an alcohol with a sulfonyl chloride by: (i) -

Methanesulfonyl Chloride
A Publication of Reliable Methods for the Preparation of Organic Compounds Working with Hazardous Chemicals The procedures in Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices. In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red “Caution Notes” within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices. The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein. -

Enhanced Conversion Oflactose to Glycerol by Kluyveromyces Fragilis
APPLIED AND ENVIRONMENTAL MICROBIOLOGY, Mar. 1989, p. 573-578 Vol. 55, No. 3 0099-2240/89/030573-06$02.00/0 Copyright C) 1989, American Society for Microbiology Enhanced Conversion of Lactose to Glycerol by Kluyveromyces fragilis Utilizing Whey Permeate as a Substrate WHEAMEI JENQ,1 RAY A. SPECKMAN,' RICHARD E. CRANG,2* AND MARVIN P. STEINBERG1 Department of Food Science, 1304 West Pennsylvania Avenue,' and School of Life Sciences, 505 South Goodwin Avenue,2 University ofIllinois, Urbana, Illinois 61801 Received 6 June 1988/Accepted 12 December 1988 Kluyveromycesfragilis (CBS 397) is a nonhalophilic yeast which is capable of lactose utilization from whey permeate and high glycerol production under anaerobic growth conditions. However, the optimum yields of glycerol (11.6 mg/ml of whey permeate medium) obtained in this study occurred only in the presence of 1% Na2SO3 as a steering agent. The use of other concentrations of Na2SO3, as well as 5% NaCl and 1% ascorbic acid, had no or detrimental effects on cell growth, lactose utilization, and glycerol production. Glycerol yields were greater in cultures grown from a light inoculum of K. fragilis than in cultures in which a resuspended mass of cells was introduced into the medium. The results of this study suggest that this strain of K. fragilis may be useful commercially in the utilization of cheese whey lactose and the concomitant production of glycerol. Cheese whey represents a commercial by-product gener- troleum derivatives, which is less expensive than processing ated in such massive quantities that its safe disposal is a by sugar fermentation. major problem for many municipal sewage treatment plants. -

Radical Approaches to Alangium and Mitragyna Alkaloids
Radical Approaches to Alangium and Mitragyna Alkaloids A Thesis Submitted for a PhD University of York Department of Chemistry 2010 Matthew James Palframan Abstract The work presented in this thesis has focused on the development of novel and concise syntheses of Alangium and Mitragyna alkaloids, and especial approaches towards (±)-protoemetinol (a), which is a key precursor of a range of Alangium alkaloids such as psychotrine (b) and deoxytubulosine (c). The approaches include the use of a key radical cyclisation to form the tri-cyclic core. O O O N N N O O O H H H H H H O N NH N Protoemetinol OH HO a Psychotrine Deoxytubulosine b c Chapter 1 gives a general overview of radical chemistry and it focuses on the application of radical intermolecular and intramolecular reactions in synthesis. Consideration is given to the mediator of radical reactions from the classic organotin reagents, to more recently developed alternative hydrides. An overview of previous synthetic approaches to a range of Alangium and Mitragyna alkaloids is then explored. Chapter 2 follows on from previous work within our group, involving the use of phosphorus hydride radical addition reactions, to alkenes or dienes, followed by a subsequent Horner-Wadsworth-Emmons reaction. It was expected that the tri-cyclic core of the Alangium alkaloids could be prepared by cyclisation of a 1,7-diene, using a phosphorus hydride to afford the phosphonate or phosphonothioate, however this approach was unsuccessful and it highlighted some limitations of the methodology. Chapter 3 explores the radical and ionic chemistry of a range of silanes. -

Hydrophilic Thin Coating and Method of Manufacturing the Same
Europaisches Patentamt European Patent Office © Publication number: 0 599 150 A1 Office europeen des brevets EUROPEAN PATENT APPLICATION © Application number: 93118306.5 int. CIA B05D 1/18, C03C 17/30, C08J 7/04 @ Date of filing: 11.11.93 ® Priority: 12.11.92 JP 302124/92 © Applicant: MATSUSHITA ELECTRIC INDUSTRIAL Co., Ltd. @ Date of publication of application: 1006-banchi, Oaza-Kadoma 01.06.94 Bulletin 94/22 Kadoma-shi, Osaka(JP) © Designated Contracting States: @ Inventor: Ohtake, Tadashi DE FR GB Kawakitanaka-machi 30-15 Neyagawa-shi, Osaka 572(JP) Inventor: Mino, Norihisa Senrioka Higashi 4-6-8-806 Settsu-shi, Osaka 566(JP) Inventor: Ogawa, Kazufumi Aoyama 2-3-50 Nara-shi, Nara 630(JP) © Representative: VOSSIUS & PARTNER Siebertstrasse 4 D-81675 Munchen (DE) © Hydrophilic thin coating and method of manufacturing the same. © The invention relates to a hydrophilic thin film and a method of manufacturing the same in which a hydrophilic thin film 4 is formed by incorporating or fixing molecules comprising hydrophilic groups to a chemically adsorbed film on the surface of a substrate 1 . 7\ oj— Cl O I OH 2 ^4 $ -0— Si — 0 Si — 0 Si- -o- -Si-0- I I I I 0 0 0 0 — > 7> , , , 1 , , r-i-. i ///////////// X FIG . Rank Xerox (UK) Business Services (3. 10/3.09/3.3.4) EP 0 599 150 A1 The invention relates to a hydrophilic thin film and a method of manufacturing the same. More specifically, the invention relates to a hydrophilic thin film and its method of manufacture, in which molecules comprising hydrophilic groups are incorporated or chemically bonded to the surface of a chemically adsorbed film on a substrate surface. -

Sodium Chlorite Neutralization
® Basic Chemicals Sodium Chlorite Neutralization Introduction that this reaction is exothermic and liberates a If sodium chlorite is spilled or becomes a waste, significant amount of heat (H). it must be disposed of in accordance with local, state, and Federal regulations by a NPDES NaClO2 + 2Na2SO3 2Na2SO4 + NaCl permitted out-fall or in a permitted hazardous 90.45g + 2(126.04g) 2(142.04g) + 58.44g waste treatment, storage, and disposal facility. H = -168 kcal/mole NaClO2 Due to the reactivity of sodium chlorite, neutralization for disposal purposes should be For example, when starting with a 5% NaClO2 avoided whenever possible. Where permitted, solution, the heat generated from this reaction the preferred method for handling sodium could theoretically raise the temperature of the chlorite spills and waste is by dilution, as solution by 81C (146F). Adequate dilution, discussed in the OxyChem Safety Data Sheet thorough mixing and a slow rate of reaction are (SDS) for sodium chlorite in Section 6, important factors in controlling the temperature (Accidental Release Measures). Sodium chlorite increase (T). neutralization procedures must be carried out only by properly trained personnel wearing Procedure appropriate protective equipment. The complete neutralization procedure involves three sequential steps: dilution, chlorite Reaction Considerations reduction, and alkali neutralization. The dilution If a specific situation requires sodium chlorite to step lowers the strength of the sodium chlorite be neutralized, the chlorite must first be reduced solution to 5% or less; the reduction step reacts by a reaction with sodium sulfite. The use of the diluted chlorite solution with sodium sulfite to sodium sulfite is recommended over other produce a sulfate solution, and the neutralization reducing agents such as sodium thiosulfate step reduces the pH of the alkaline sulfate (Na2S2O3), sodium bisulfite (NaHSO3), and solution from approximately 12 to 4-5. -

덴드리틱 벤질 클로라이드의 효율적인 합성 Facile Synthesis of Dendritic Benzyl Chlorides from Their Alcohols W
Polymer(Korea), Vol. 31, No. 5, pp 417-421, 2007 덴드리틱 벤질 클로라이드의 효율적인 합성 이재욱F,7ㆍ한승철Fㆍ김희주ㆍ김정환ㆍ이언엽ㆍ김병기ㆍ성새름ㆍ강화신ㆍ김지현FFㆍ허도성FFF 동아대학교 화학과, F동아대학교 의생명과학과, FF동국대학교 생명화학공학과, FFF인제대학교 의생명화학과 (2007년 5월 9일 접수, 2007년 6월 29일 채택) Facile Synthesis of Dendritic Benzyl Chlorides from Their Alcohols with Methanesulfonyl Chloride/Et3N Jae Wook Lee*,7, Seung Choul Han*, Hee Joo Kim, Jung Hwan Kim, Un Yup Lee, Byoung-Ki Kim, Sae Reum Sung, Hwa-Shin Kang, Ji Hyeon Kim**, and Do-Sung Huh*** Department of Chemistry, Dong-A University, Hadan-2-dong, Busan 604-714, Korea *Department of Medical Bioscience, Dong-A University, Hadan-2-dong, Busan 604-714, Korea **Department of Chemical & Biochemical Engineering, Dongguk University, Seoul 100-715, Korea ***Department of Biomedicinal Chemistry, Inje University, Gim Hai 621-749, Korea (Received May 9, 2007; Accepted June 29, 2007) 초록: 덴드리틱 벤질 알코올을 트리에틸아민과 메탄술포닐클로라이드와 반응시켜서, 덴드리틱 벤질 클로라이드 의 효율적인 합성이 이루어졌다. 이 반응은 히드록시기의 메실화 반응과 염소화 반응의 2단계 반응으로 이루어지 는데, 중간체의 분리없이 한 반응 용기내에서 반응이 진행되는 경제적인 방법이다. Abstract: A successful rapid synthesis of dendritic benzyl chlorides from dendritic benzyl alcohols using methanesulfonyl chloride/Et3N as activating agents was described. In this method, each dendritic benzyl chloride can be prepared in one pot: no isolation of intermediate mesylated dendrons is required. The key steps in the syntheses of dendritic benzyl chlorides were the mesylation of the hydroxymethyl group followed by the chlorination by in-situ generated triethylammonium chloride. Keywords: benzylic alcohol, chlorination. Introduction vation and coupling steps in high yield. In addition, the coupling step must be very efficient to enable complete Dendrimers represent a novel type of polymeric material reaction. -

R. B. Woodward Precipitation of Barium in the Copper-Tin Group Of
HETEROCYCLES, Vol. 7, No. 1. 1977 R. B. Woodward Precipitation of barium in the copper-tin group of qualitative analysis. W.J.Hal1 and RBW, -Ind. Eng. Chem., Anal. Ed., 6, 478 (1934). A new pressure regulator for vacuum distillation. R.L.Emerson and RBW, g.,2, 347 (1937). The Staling of coffee. II. S.C.Prescott, R.L.Emerson, RBW, and R. Heggie, Food Re- --search, 2, 165 (1937). Pyrolysis of organomagnesium compounds. I. A new agent for the reduction of bemophenone. D.B.Clapp, and RBW, 1. ---Am. Chem. Soc., -60, 1019 (1938). The direct introduction of the angular methyl group. RBW, Ibid., 62, 1208 (1940). Experiments on the synthesis of oestrone. I. 2-(8-phenylethy1)-furans as components in the diene synthesis. RBW, Ibid., 62, 1478 (1940). The formation of Reissert's compounds in non-aqueous media. RBW, Ibid., 62, 1626 (1940). A new optically active reagent for carbonyl compounds. The resolution of -dl-camphor. RBW, T. P. Kohman, and G. C. Harris, Ibid., 63, lu) (1941). The isolation and properties of 1, 1-dineopentylethylene, a component of triisobutylene. P.D.Bartlett, G.L. Fraser, and RBW, x.,3, 495 (1941). Structure and absorption spectra of a,p-unsaturated ketones. RBW, Ibid., 3, 1123 (1941). 5 Structure and absorption spectra. II. 3-Acetoxy-A -(6)-~cholestene-7-carboxylicacid. RBW, and A.F.Clifiord, -Ibid., -63, 2727 (1941). The structure of cantharidine and the synthesis of desoxycantharidine. RLW, and R.B. Loftfield, Ibid., 3, 3167 (1941). -t-Butyllithium. P.D.Bardett, C .G.Swain, and RBW, --Ibid., 63, 3229 (1941). -

Synthesis of Tosyl- and Nosyl-Ended Polyisobutylenes with High Extent of Functionalities: the Effect of Reaction Conditions
polymers Article Synthesis of Tosyl- and Nosyl-Ended Polyisobutylenes with High Extent of Functionalities: The Effect of Reaction Conditions Balázs Pásztói 1,2,* , Tobias M. Trötschler 3,4,5, Ákos Szabó 1, Györgyi Szarka 1, Benjamin Kerscher 3,4 , Rolf Mülhaupt 3,4,5,* and Béla Iván 1,* 1 Polymer Chemistry Research Group, Institute of Materials and Environment Chemistry, Research Centre for Natural Sciences, Magyar tudósok körútja 2, H-1117 Budapest, Hungary; [email protected] (Á.S.); [email protected] (G.S.) 2 George Hevesy PhD School of Chemistry, Institute of Chemistry, Faculty of Science, Eötvös Loránd University, Pázmány Péter sétány 2, H-1117 Budapest, Hungary 3 Institute for Macromolecular Chemistry, University of Freiburg, Stefan-Meier-Str. 31, D-79104 Freiburg, Germany; [email protected] (T.M.T.); [email protected] (B.K.) 4 Freiburg Materials Research Center, University of Freiburg, Stefan-Meier-Str. 21, D-79104 Freiburg, Germany 5 Freiburg Center for Interactive Materials and Bioinspired Technologies (FIT), University of Freiburg, Georges-Köhler-Allee 105, D-79110 Freiburg, Germany * Correspondence: [email protected] (B.P.); [email protected] (R.M.); [email protected] (B.I.) Received: 7 October 2020; Accepted: 24 October 2020; Published: 28 October 2020 Abstract: Endfunctional polymers possess significant industrial and scientific importance. Sulfonyl endgroups, such as tosyl and nosyl endfunctionalities, due their ease of substitution are highly desired for a variety of polymer structures. The sulfonylation of hydroxyl-terminated polyisobutylene (PIB-OH), a chemically and thermally stable, biocompatible, fully saturated polymer, with tosyl chloride (TsCl) and nosyl chloride (NsCl) is presented in this study. -

University of Cincinnati
UNIVERSITY OF CINCINNATI DATE: July 5, 2002 I, Dinesh Kumar Palaniswamy , hereby submit this as part of the requirements for the degree of: Master of Science in: Environmental Engineering It is entitled: Electrochemical Reduction of 2,4,6-Trinitrotoluene Approved by: Dr.George Sorial Dr.Dionysios Dionysiou Dr.Makram Suidan ELECTROCHEMICAL REDUCTION OF 2,4,6 -TRINITROTOLUENE A thesis submitted to the Division of Research and Advanced Studies of the University of Cincinnati in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE in the Department of Civil and Environmental Engineering of the college of Engineering 2002 by Dinesh Kumar Palaniswamy B.E., Civil Engineering, P.S.G College of Technology, Coimbatore, 1998 Committee Chair: Dr. George Sorial ABSTRACT 2,4,6-trinitrotoluene (TNT) is a major constituent of munitions contaminated wastewater. This research aims at studying the efficiency of electrochemical processes in reducing TNT. A laboratory scale reactor was designed and developed to electrochemically reduce TNT in simulated munition wastewater. Experiments simulating batch conditions were first conducted on the laboratory scale reactor to study the effect of various parameters including applied current, type of electrolyte and the molar concentration of the electrolyte in the feed solution on the reduction kinetics of TNT. The results showed that the reduction rates of TNT increased with an increase in applied current and molar concentration of electrolyte in feed. The rates of reduction leveled off at higher currents (250 & 300 mA). Mass transfer limitations were speculated for this flattening of rate constants at higher currents. Experimental studies were conducted with two types of electrolyte in the feed- sodium sulfate (Na2SO4) and lithium sulfate (Li2SO4), and the results indicated that there was no significant difference in the reduction rates for TNT. -
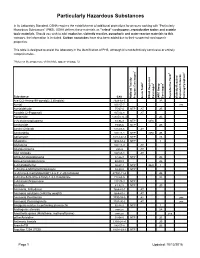
Particularly Hazardous Substances
Particularly Hazardous Substances In its Laboratory Standard, OSHA requires the establishment of additional protections for persons working with "Particularly Hazardous Substances" (PHS). OSHA defines these materials as "select" carcinogens, reproductive toxins and acutely toxic materials. Should you wish to add: explosive, violently reactive, pyrophoric and water-reactve materials to this category, the information is included. Carbon nanotubes have also been added due to their suspected carcinogenic properties. This table is designed to assist the laboratory in the identification of PHS, although it is not definitively conclusive or entirely comprehensive. *Notes on the proper use of this table appear on page 12. 1 6 5 2 3 4 Substance CAS National Toxicity National Program Carcinogen Toxin Acute Regulated OSHA Carcinogen Group IARC Carcinogen Toxin Reproductive Violently Reactive/ Explosive/Peroxide Forming/Pyrophoric A-a-C(2-Amino-9H-pyrido[2,3,b]indole) 2648-68-5 2B Acetal 105-57-7 yes Acetaldehyde 75-07-0 NTP AT 2B Acrolein (2-Propenal) 107-02-8 AT Acetamide 126850-14-4 2B 2-Acetylaminofluorene 53-96-3 NTP ORC Acrylamide 79-06-6 NTP 2B Acrylyl Chloride 814-68-6 AT Acrylonitrile 107-13-1 NTP ORC 2B Adriamycin 23214-92-8 NTP 2A Aflatoxins 1402-68-2 NTP 1 Allylamine 107-11-9 AT Alkylaluminums varies AT Allyl Chloride 107-05-1 AT ortho-Aminoazotoluene 97-56-3 NTP 2B para-aminoazobenzene 60-09-3 2B 4-Aminobiphenyl 92-67-1 NTP ORC 1 1-Amino-2-Methylanthraquinone 82-28-0 NTP (2-Amino-6-methyldipyrido[1,2-a:3’,2’-d]imidazole) 67730-11-4 2B -

Sulfur Dioxide and Some Sulfites, Bisulfites and Metabisulfites
SULFUR DIOXIDE AND SOME SULFITES, BISULFITES AND METABISULFITES 1. Exposure Data 1.1 Chemical and physical data 1.1.1 Synonyms and structural and molecular data Sulfr dioxi Chem. Abstr. Serv Reg. No.: 7446-09-5 Replaced CAS Nos.: 8014-94-6; 12396-99-5; 83008-56-4; 89125-89-3 Chem. Abstr. Name; Sulfur dioxide IUPAC Systematic Name: Sulfur dioxide Synonyms: Sulfurous acid anhydride; sulfurous anhydride; sulfurous oxide; sulfur oxide (S02); sulfur superoxide; sulphur dioxide 0=8=0 S02 MoL. wt: 64.07 Sodium sulfte Chem. Abstr. Serv Reg. No.: 7757-83-7 Altemate CAS No.: 10579-83-6 Replaced CAS No.: 68135-69-3 Chem. Abstr. Name: Sulfurous acid, di sodium salt IUPAC Systematic Name: Sulfurous acid, disodium salt Synonyms: Anhydrous sodium sulfite; disodium sulfite; sodium sulphite o 1/ Na · 0 - 8 - 0 · Na Na2S0J MoL. wt: 126.04 Sodium bisulfe Chem. Abstr. Serv Reg. No.: 7631-90-5 Replaced CAS Nos.: 57414-01-4; 69098-86-8; 89830-27-3; 91829-63-9 Chem. Abstr. Name: Sulfurous acid, monosodium salt IUPAC Systematic Name: Sulfurous acid, monosodium salt -131- 132 lARe MONOGRAPHS VOLUME 54 Synonyms: Hydrogen sulfite sodium; monosodium sulfite; sodium acid sulfite; sodium bisulphite; sodium hydrogen sulfite; sodium sulfite (NaHS03) o Il HO - S - a · Na NaHS03 MoL. wt: 104.06 Sodium metabisulfte Chem. Abstr. Serv Reg. No.: 7681-57-4 Altemate CAS No.: 7757-74-6 Replaced CAS No.: 15771-29-6 Chem. Abstr. Name: Disulfurous acid, disodium salt IUPAC Systematic Name: Pyrosulfurous acid, disodium salt Synonyms: Disodium disulfite; disodium metabisulfite; disodium pyrosulfite; sodium disulfite; sodium metabisulphite; sodium pyrosulfite oIl Il0 Na · 0- S - a - S - a · Na .Na2S20S MoL.