Dystonia Precision Panel Overview Indications Clinical Utility
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Enzymatic Encoding Methods for Efficient Synthesis Of
(19) TZZ__T (11) EP 1 957 644 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C12N 15/10 (2006.01) C12Q 1/68 (2006.01) 01.12.2010 Bulletin 2010/48 C40B 40/06 (2006.01) C40B 50/06 (2006.01) (21) Application number: 06818144.5 (86) International application number: PCT/DK2006/000685 (22) Date of filing: 01.12.2006 (87) International publication number: WO 2007/062664 (07.06.2007 Gazette 2007/23) (54) ENZYMATIC ENCODING METHODS FOR EFFICIENT SYNTHESIS OF LARGE LIBRARIES ENZYMVERMITTELNDE KODIERUNGSMETHODEN FÜR EINE EFFIZIENTE SYNTHESE VON GROSSEN BIBLIOTHEKEN PROCEDES DE CODAGE ENZYMATIQUE DESTINES A LA SYNTHESE EFFICACE DE BIBLIOTHEQUES IMPORTANTES (84) Designated Contracting States: • GOLDBECH, Anne AT BE BG CH CY CZ DE DK EE ES FI FR GB GR DK-2200 Copenhagen N (DK) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • DE LEON, Daen SK TR DK-2300 Copenhagen S (DK) Designated Extension States: • KALDOR, Ditte Kievsmose AL BA HR MK RS DK-2880 Bagsvaerd (DK) • SLØK, Frank Abilgaard (30) Priority: 01.12.2005 DK 200501704 DK-3450 Allerød (DK) 02.12.2005 US 741490 P • HUSEMOEN, Birgitte Nystrup DK-2500 Valby (DK) (43) Date of publication of application: • DOLBERG, Johannes 20.08.2008 Bulletin 2008/34 DK-1674 Copenhagen V (DK) • JENSEN, Kim Birkebæk (73) Proprietor: Nuevolution A/S DK-2610 Rødovre (DK) 2100 Copenhagen 0 (DK) • PETERSEN, Lene DK-2100 Copenhagen Ø (DK) (72) Inventors: • NØRREGAARD-MADSEN, Mads • FRANCH, Thomas DK-3460 Birkerød (DK) DK-3070 Snekkersten (DK) • GODSKESEN, -

Generated by SRI International Pathway Tools Version 25.0, Authors S
An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000238675-HmpCyc: Bacillus smithii 7_3_47FAA Cellular Overview Connections between pathways are omitted for legibility. -

Circuits Regulating Superoxide and Nitric Oxide
antioxidants Article Circuits Regulating Superoxide and Nitric Oxide Production and Neutralization in Different Cell Types: Expression of Participating Genes and Changes Induced by Ionizing Radiation 1,2, 1,2, 1 1,2, Patryk Bil y, Sylwia Ciesielska y, Roman Jaksik and Joanna Rzeszowska-Wolny * 1 Department of Systems Biology and Engineering, Faculty of Automatic Control, Electronics and Computer Science, Silesian University of Technology, 44-100 Gliwice, Poland; [email protected] (P.B.); [email protected] (S.C.); [email protected] (R.J.) 2 Biotechnology Centre, Silesian University of Technology, 44-100 Gliwice, Poland * Correspondence: [email protected] These authors contributed equally. y Received: 30 June 2020; Accepted: 29 July 2020; Published: 3 August 2020 Abstract: Superoxide radicals, together with nitric oxide (NO), determine the oxidative status of cells, which use different pathways to control their levels in response to stressing conditions. Using gene expression data available in the Cancer Cell Line Encyclopedia and microarray results, we compared the expression of genes engaged in pathways controlling reactive oxygen species and NO production, neutralization, and changes in response to the exposure of cells to ionizing radiation (IR) in human cancer cell lines originating from different tissues. The expression of NADPH oxidases and NO synthases that participate in superoxide radical and NO production was low in all cell types. Superoxide dismutase, glutathione peroxidase, thioredoxin, and peroxiredoxins participating in radical neutralization showed high expression in nearly all cell types. Some enzymes that may indirectly influence superoxide radical and NO levels showed tissue-specific expression and differences in response to IR. -

Transcriptional Analysis of Sodium Valproate in a Serotonergic Cell Line Reveals Gene Regulation Through Both HDAC Inhibition-Dependent and Independent Mechanisms
bioRxiv preprint doi: https://doi.org/10.1101/837732; this version posted November 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Transcriptional analysis of sodium valproate in a serotonergic cell line reveals gene regulation through both HDAC inhibition-dependent and independent mechanisms Priyanka Sinha1,2, Simone Cree1,2, Allison L. Miller1,2, John F. Pearson1,2,3, Martin A. Kennedy1,2. 1Gene Structure and Function Laboratory, Department of Pathology and Biomedical Science, University of Otago, Christchurch, New Zealand. 2Carney Centre for Pharmacogenomics, University of Otago, Christchurch, New Zealand. 3Biostatistics and Computational Biology Unit, University of Otago, Christchurch, New Zealand. Correspondence to: Prof. M. A. Kennedy Department of Pathology and Biomedical Science University of Otago, Christchurch Christchurch, New Zealand Email: [email protected] Keywords: RNA-Seq, NanoString, lithium, valproate, HDAC inhibitor, mood stabilizer 1 bioRxiv preprint doi: https://doi.org/10.1101/837732; this version posted November 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract Sodium valproate (VPA) is a histone deacetylase (HDAC) inhibitor, widely prescribed in the treatment of bipolar disorder, and yet the precise modes of therapeutic action for this drug are not fully understood. -

Gene Transfer As a Potential Treatment for Tetralujdrobiopterin Deficient States
Gene Transfer as a Potential Treatment for Tetralujdrobiopterin Deficient States Rickard F oxton Division of Neurochemistry Department of Molecular Neuroscience Institute of Neurology University College London Submitted November 2006 Funded by Brain Research Trust Thesis submitted for the degree of Doctor of Philosophy, University of London. I, Richard Hartas Foxton, confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis.' 1 UMI Number: U592813 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. Dissertation Publishing UMI U592813 Published by ProQuest LLC 2013. Copyright in the Dissertation held by the Author. Microform Edition © ProQuest LLC. All rights reserved. This work is protected against unauthorized copying under Title 17, United States Code. ProQuest LLC 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106-1346 ABSTRACT Tetrahydrobiopterin (BH4) is an essential cofactor for dopamine (DA), noradrenaline (NA), serotonin and nitric oxide (NO) synthesis in the brain. Inborn errors of BH4 metabolism including GTP cyclohydrolase 1 (GTP-CH) deficiency are debilitating diseases in which BH4, DA, 5-HT and NO metabolism are impaired. Current treatment for these disorders is typically monoamine replacement +/- BH4. Whilst correction of the primary defect is the ideal, BH4 treatment is problematic as it is expensive and inefficacious. -

SET-Induced Calcium Signaling and MAPK/ERK Pathway Activation Mediate Dendritic Cell-Like Differentiation of U937 Cells
Leukemia (2005) 19, 1439–1445 & 2005 Nature Publishing Group All rights reserved 0887-6924/05 $30.00 www.nature.com/leu SET-induced calcium signaling and MAPK/ERK pathway activation mediate dendritic cell-like differentiation of U937 cells A Kandilci1 and GC Grosveld1 1Department of Genetics and Tumor Cell Biology, Mail Stop 331, St Jude Children’s Research Hospital, 332 N. Lauderdale, Memphis, TN 38105, USA Human SET, a target of chromosomal translocation in human G1/S transition by allowing cyclin E–CDK2 activity in the leukemia encodes a highly conserved, ubiquitously expressed, presence of p21.11 Second, SET interacts with cyclin B–CDK1.19 nuclear phosphoprotein. SET mediates many functions includ- ing chromatin remodeling, transcription, apoptosis and cell Overexpression of SET inhibits cyclin B-CDK1 activity, which in cycle control. We report that overexpression of SET directs turn, blocks the G2/M transition; this finding suggests a negative 13 differentiation of the human promonocytic cell line U937 along regulatory role for SET in G2/M transition. Overexpression of the dendritic cell (DC) pathway, as cells display typical cell division autoantigen-1 (CDA1), another member of the morphologic changes associated with DC fate and express NAP/SET family, inhibits proliferation and decreases bromo- the DC surface markers CD11b and CD86. Differentiation occurs deoxyuridine uptake in HeLa cells.20 Acidic and basic domains via a calcium-dependent mechanism involving the CaMKII and 20 MAPK/ERK pathways. Similar responses are elicited by inter- of CDA1 show 40% identity and 68% similarity to SET. feron-c (IFN-c) treatment with the distinction that IFN-c signaling We have recently shown that overexpression of SET in the activates the DNA-binding activity of STAT1 whereas SET human promonocytic cell line U937 causes G0/G1 arrest and overexpression does not. -

Biochemical and Functional Characterization of Plasmodium
Kümpornsin et al. Malaria Journal 2014, 13:150 http://www.malariajournal.com/content/13/1/150 RESEARCH Open Access Biochemical and functional characterization of Plasmodium falciparum GTP cyclohydrolase I Krittikorn Kümpornsin1†, Namfon Kotanan1†, Pornpimol Chobson1,4, Theerarat Kochakarn1, Piyaporn Jirawatcharadech1, Peera Jaru-ampornpan2, Yongyuth Yuthavong2 and Thanat Chookajorn1,3* Abstract Background: Antifolates are currently in clinical use for malaria preventive therapy and treatment. The drugs kill the parasites by targeting the enzymes in the de novo folate pathway. The use of antifolates has now been limited by the spread of drug-resistant mutations. GTP cyclohydrolase I (GCH1) is the first and the rate-limiting enzyme in the folate pathway. The amplification of the gch1 gene found in certain Plasmodium falciparum isolates can cause antifolate resistance and influence the course of antifolate resistance evolution. These findings showed the importance of P. falciparum GCH1 in drug resistance intervention. However, little is known about P. falciparum GCH1 in terms of kinetic parameters and functional assays, precluding the opportunity to obtain the key information on its catalytic reaction and to eventually develop this enzyme as a drug target. Methods: Plasmodium falciparum GCH1 was cloned and expressed in bacteria. Enzymatic activity was determined by the measurement of fluorescent converted neopterin with assay validation by using mutant and GTP analogue. The genetic complementation study was performed in ΔfolE bacteria to functionally identify the residues and domains of P. falciparum GCH1 required for its enzymatic activity. Plasmodial GCH1 sequences were aligned and structurally modeled to reveal conserved catalytic residues. Results: Kinetic parameters and optimal conditions for enzymatic reactions were determined by the fluorescence-based assay. -

Proteasome Biology: Chemistry and Bioengineering Insights
polymers Review Proteasome Biology: Chemistry and Bioengineering Insights Lucia Raˇcková * and Erika Csekes Centre of Experimental Medicine, Institute of Experimental Pharmacology and Toxicology, Slovak Academy of Sciences, Dúbravská cesta 9, 841 04 Bratislava, Slovakia; [email protected] * Correspondence: [email protected] or [email protected] Received: 28 September 2020; Accepted: 23 November 2020; Published: 4 December 2020 Abstract: Proteasomal degradation provides the crucial machinery for maintaining cellular proteostasis. The biological origins of modulation or impairment of the function of proteasomal complexes may include changes in gene expression of their subunits, ubiquitin mutation, or indirect mechanisms arising from the overall impairment of proteostasis. However, changes in the physico-chemical characteristics of the cellular environment might also meaningfully contribute to altered performance. This review summarizes the effects of physicochemical factors in the cell, such as pH, temperature fluctuations, and reactions with the products of oxidative metabolism, on the function of the proteasome. Furthermore, evidence of the direct interaction of proteasomal complexes with protein aggregates is compared against the knowledge obtained from immobilization biotechnologies. In this regard, factors such as the structures of the natural polymeric scaffolds in the cells, their content of reactive groups or the sequestration of metal ions, and processes at the interface, are discussed here with regard to their -

GCH1 Gene GTP Cyclohydrolase 1
GCH1 gene GTP cyclohydrolase 1 Normal Function The GCH1 gene provides instructions for making an enzyme called GTP cyclohydrolase 1. This enzyme is involved in the first of three steps in the production of a molecule called tetrahydrobiopterin (BH4). Other enzymes help carry out the second and third steps in this process. Tetrahydrobiopterin plays a critical role in processing several protein building blocks ( amino acids) in the body. For example, it works with the enzyme phenylalanine hydroxylase to convert an amino acid called phenylalanine into another amino acid, tyrosine. Tetrahydrobiopterin is also involved in reactions that produce chemicals called neurotransmitters, which transmit signals between nerve cells in the brain. Specifically, tetrahydrobiopterin is involved in the production of two neurotransmitters called dopamine and serotonin. Among their many functions, dopamine transmits signals within the brain to produce smooth physical movements, and serotonin regulates mood, emotion, sleep, and appetite. Because it helps enzymes carry out chemical reactions, tetrahydrobiopterin is known as a cofactor. Health Conditions Related to Genetic Changes Dopa-responsive dystonia More than 140 mutations in the GCH1 gene have been found to cause dopa-responsive dystonia. This condition is characterized by a pattern of involuntary muscle contractions ( dystonia), tremors, and other uncontrolled movements and usually responds to treatment with a medication called L-Dopa. Dopa-responsive dystonia results when one copy of the GCH1 gene is mutated in each cell. Most GCH1 gene mutations that cause this condition change single amino acids in the GTP cyclohydrolase 1 enzyme. Researchers believe that the abnormal enzyme may interfere with the activity of the normal version of GTP cyclohydrolase 1 that is produced from the copy of the gene with no mutation. -
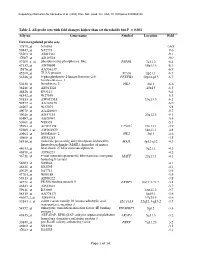
Table 2. All Probe Sets with Fold Changes Higher Than Set Thresholds but P > 0.001 Affy No
Supporting information for Sanoudou et al. (2003) Proc. Natl. Acad. Sci. USA, 10.1073/pnas.0330960100 Table 2. All probe sets with fold changes higher than set thresholds but P > 0.001 Affy no. Gene name Symbol Location Fold Down-regulated probe sets 47879_at N46863 -16.9 59447_at N52773 -9.6 55503_at AI085361 -9.1 47087_at AI310524 -9.0 37209_g_at phosphoserine phosphatase-like PSPHL 7q11.2 -8.4 47137_at AI479899 19p13.3 -8.1 45876_at AA536137 -6.9 45260_at TU3A protein TU3A 3p21.1 -6.7 56246_at 6-phosphofructo-2-kinase/fructose-2,6- PFKFB3 10p14-p15 -6.7 bisphosphatase 3 50230_at hexokinase 2 HK2 2p13 -6.6 38248_at AB011124 20p13 -6.5 44426_at R93141 -6.4 48542_at W27559 -6.3 53813_at AW051518 13q13.3 -6.1 59577_at AA243670 -6.0 46907_at W37075 -5.8 49078_at AA424983 -5.7 49026_at AI357153 20p12.3 -5.4 53487_at AI670947 -5.4 50161_at N39328 -5.1 35994_at AC002398 F25965 19q13.1 -4.9 52285_f_at AW002970 18p11.1 -4.8 40964_at hexokinase 2 HK2 2p13 -4.6 49806_at AI932283 -4.5 58918_at molecule possessing ankyrin repeats induced by MAIL 3p12-q12 -4.3 lipopolysaccharide (MAIL), homolog of mouse 44633_at heat shock 27 kDa associated protein 3q21.1 -4.3 46858_at AI796221 -4.2 36711_at v-maf musculoaponeurotic fibrosarcoma oncogene MAFF 22q13.1 -4.1 homolog F (avian) 54683_at N49844 -4.1 46621_at N32595 -4.1 49629_at N47713 -3.9 47703_at W89189 -3.9 59313_at AI598222 -3.8 34721_at FK506 binding protein 5 FKBP5 6p21.3-21.2 -3.8 46843_at AI632621 -3.7 59611_at R53069 16p11.2 -3.7 58315_at AA778171 3p25.1 -3.6 46607_f_at AI885018 17q25.3 -3.6 33143_s_at solute carrier family 16 (monocarboxylic acid SLC16A3 22q12.3-q13.2 -3.5 transporters), member 3 54152_at eukaryotic translation initiation factor 4E binding EIF4EBP1 8p12 -3.4 protein 1 43935_at ARF-GAP, RHO-GAP, ankyrin repeat and plekstrin ARAP3 5q31.3 -3.2 homology domains-containing protein 3 33849_at pre-B-cell colony-enhancing factor PBEF 7q11.23 -3.2 46902_at N92294 -3.2 47023_at N25555 -3.1 Page 1 of 14 Supporting information for Sanoudou et al. -

1 SUPPLEMENTARY INFORMATION 1 2 an Archaeal Symbiont
1 SUPPLEMENTARY INFORMATION 2 3 An archaeal symbiont-host association from the deep terrestrial subsurface 4 5 Katrin Schwank1, Till L. V. Bornemann1, Nina Dombrowski2, Anja Spang2,3, Jillian F. Banfield4 and 6 Alexander J. Probst1* 7 8 9 1 Group for Aquatic Microbial Ecology (GAME), Biofilm Center, Department of Chemistry, 10 University of Duisburg-Essen, Germany 11 2Department of Marine Microbiology and Biogeochemistry (MMB), Royal Netherlands Institute 12 for Sea Research (NIOZ), and Utrecht University, Den Burg, Netherlands 13 3Department of Cell- and Molecular Biology, Science for Life Laboratory, Uppsala University, SE- 14 75123, Uppsala, Sweden 15 4 Department for Earth and Planetary Sciences, University of California, Berkeley, USA 16 17 *to whom the correspondence should be addressed: 18 Alexander J. Probst 19 University of Duisburg-Essen 20 Universitätsstrasse 4 21 45141 Essen 22 [email protected] 23 24 25 Content: 26 1. Supplementary Methods 27 2. Supplementary Tables 28 3. Supplementary Figure 29 4. References 30 31 32 33 1. Supplementary methods 34 Phylogenetic analysis 35 Hmm profiles of the 37 maker genes used by the phylosift v1.01 software [1, 2] were queried 36 against a representative set of archaeal genomes from NCBI (182) and the GOLD database (3) as 37 well as against the huberarchaeal population genome (for details on genomes please see 38 Supplementary Data file 1) using the phylosift search mode [1, 2]. Protein sequences 39 corresponding to each of the maker genes were subsequently aligned using mafft-linsi v7.407 [3] 40 and trimmed with BMGE-1.12 (parameters: -t AA -m BLOSUM30 -h 0.55) [4]. -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S.