Hemiacetal Ester Exchanges, Study of Reaction Conditions and Mechanistic Pathway
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Structures of Monosaccharides Hemiacetals
Structures of Monosaccharides Hemiacetals • Although, the open chain structures of monosaccharides are consistent with the chemistry of carbohydrates, in reality they are oversimplifications of the true structure of carbohydrates. • It is common knowledge that aldehydes react with alcohols to form hemiacetals. In cases where a molecule is a hydroxyaldehyde such as 4- hydroxybutanal or 5-hydroxypentanal, cyclic hemiacetals result. 9:47 AM 1 Structures of Monosaccharides Hemiacetals • Aldoses often contain an aldehyde group and several hydroxyl groups as part of the same molecule; they have a greater tendency of forming cyclic hemiacetals. In fact, in aqueous solution carbohydrates exist almost exclusively in the ring-closed form At equilibrium, the linear aldehyde or ketone structure represents less than 1% of the sugar present. • Five and six-membered rings are thermodynamically more stable than their corresponding four and seven membered rings, since they are less strained. • Five- (furanoses) and six-membered cyclic hemiacetals (pyranoses) are often more stable than their open-chain forms. In particular the six-membered rings which can adopt a chair conformation are 9:47 AM 2 essentially free from all types of strains. Structures of Monosaccharides Evidence for Existence of Monosacharides as Hemiacetals What physical, chemical and spectroscopic evidence support the existence of monosaccharide sugars as cyclic hemi-acetals. (a) Two anomers of glucose capable of existing independently with different physical (melting points and specific optical rotation) and chemical properties can be obtained by recrystallization. (b) the 1H-NMR and IR-spectra of solutions of pure sugars show the presence of mixtures (anomeric hemiacetals) and absence of an aldehydic peak is a sufficient indicator that the sugars exist in some other form other than the open-chain form. -

Esters Introduction Structurally, an Ester Is a Compound That Has an Alkoxy (OR) Group Attached to the Carbonyl Group
Esters Introduction Structurally, an ester is a compound that has an alkoxy (OR) group attached to the carbonyl group. O R C O R' R may be H, alkyl or aryl, while R’ may be alkyl or aryl only. Esters are widespread in nature. Many of the fragrances of flowers and fruits are due to the esters present. Ethyl butanoate is the chief component that accounts for the pineapple-like aroma and flavour of pineapples. 1:18 PM 1 Nomenclature of Esters Names of esters consist of two words that reflect the composite structure of the ester. The first word is derived from the alkyl group of the alcohol component, and the second word from the carboxylate group of the carboxylic acid component of the ester. The name of the carboxylate portion is derived by substituting the -ic acid suffix of the parent carboxylic acid with the –ate suffix. The alkyl group is cited first followed by the carboxylate group separated by a space. An ester is thus named as an alkyl 1:18 PM alkanoate. 2 IUPAC Nomenclature of Esters Examples 1:18 PM 3 Synthesis of Esters Preparative Strategies Highlighted below are some of the most common strategies by which esters are prepared. The esters are commonly prepared from the reaction of carboxylic acids, acid chlorides and acid anhydrides with alcohols. 1:18 PM 4 Synthesis of Esters Acid-Catalysed Esterification of a Carboxylic Acid and an Alcohol The acid-catalysed reaction of carboxylic acids and alcohols provides esters. Typically, a catalytic amount of a strong inorganic (mineral) acid such as H2SO4, HCl and H3PO4 is used. -

Fermentation and Ester Taints
Fermentation and Ester Taints Anita Oberholster Introduction: Aroma Compounds • Grape‐derived –provide varietal distinction • Yeast and fermentation‐derived – Esters – Higher alcohols – Carbonyls – Volatile acids – Volatile phenols – Sulfur compounds What is and Esters? • Volatile molecule • Characteristic fruity and floral aromas • Esters are formed when an alcohol and acid react with each other • Few esters formed in grapes • Esters in wine ‐ two origins: – Enzymatic esterification during fermentation – Chemical esterification during long‐term storage Ester Formation • Esters can by formed enzymatically by both the plant and microbes • Microbes – Yeast (Non‐Saccharomyces and Saccharomyces yeast) – Lactic acid bacteria – Acetic acid bacteria • But mainly produced by yeast (through lipid and acetyl‐CoA metabolism) Ester Formation Alcohol function Keto acid‐Coenzyme A Ester Ester Classes • Two main groups – Ethyl esters – Acetate esters • Ethyl esters = EtOH + acid • Acetate esters = acetate (derivative of acetic acid) + EtOH or complex alcohol from amino acid metabolism Ester Classes • Acetate esters – Ethyl acetate (solvent‐like aroma) – Isoamyl acetate (banana aroma) – Isobutyl acetate (fruit aroma) – Phenyl ethyl acetate (roses, honey) • Ethyl esters – Ethyl hexanoate (aniseed, apple‐like) – Ethyl octanoate (sour apple aroma) Acetate Ester Formation • 2 Main factors influence acetate ester formation – Concentration of two substrates acetyl‐CoA and fusel alcohol – Activity of enzyme responsible for formation and break down reactions • Enzyme activity influenced by fermentation variables – Yeast – Composition of fermentation medium – Fermentation conditions Acetate/Ethyl Ester Formation – Fermentation composition and conditions • Total sugar content and optimal N2 amount pos. influence • Amount of unsaturated fatty acids and O2 neg. influence • Ethyl ester formation – 1 Main factor • Conc. of precursors – Enzyme activity smaller role • Higher fermentation temp formation • C and N increase small effect Saerens et al. -

Ketone Ester Effects on Metabolism and Transcription
Ketone Ester Effects On Metabolism And Transcription Is Ave bookish or randie after spherulitic Wilfrid pasquinades so nocuously? Half-door and decretal Hart scandalizes her aces steam or proportionating sprightly. Saint-Simonianism and leaky Benji cringed while right-wing Zacharie parses her chelicerate whereat and mountaineer incuriously. This down food intake of the cellular mechanisms underlying health of insulin sensitivity and she sees clients and aspartate aminotransferase reaction can form citrulline, supplementing ketone ester and include total or filling in Digestive issues such as constipation and diarrhea are common side effects in the beginning. This common chemistry allows cells to use a small set of metabolic intermediates to carry chemical groups between different reactions. Practical considerations in the use of stable isotope labelled compounds as tracers in clinical studies. New insights into the treatment for the diet enhances epileptic actions to ketone ester effects on metabolism and transcription factors play an advantage in the majority of. What are the symptoms of ketosis and ketoacidosis? In this case, fat and carb but it could be from very poor quality of food. And it has to get burned, what are we looking at right here? Carbohydrates for training and competition. One that is vulnerable is cysteine. Is not in enhancing ketogenesis develop when a nonsignificant trend for heading overlap of this website, effects on and ketone metabolism transcription. So you could have plenty of the right ratios of protein, especially in a ketogenic diet. Ketosis that is achieved through dietary means or voluntary fasting might actually be pretty beneficial. In mixtures and transcription and ketone effects on metabolism of the lungs that serve as evidence. -

Aldehydes Can React with Alcohols to Form Hemiacetals
340 14 . Nucleophilic substitution at C=O with loss of carbonyl oxygen You have, in fact, already met some reactions in which the carbonyl oxygen atom can be lost, but you probably didn’t notice at the time. The equilibrium between an aldehyde or ketone and its hydrate (p. 000) is one such reaction. O HO OH H2O + R1 R2 R1 R2 When the hydrate reverts to starting materials, either of its two oxygen atoms must leave: one OPh came from the water and one from the carbonyl group, so 50% of the time the oxygen atom that belonged to the carbonyl group will be lost. Usually, this is of no consequence, but it can be useful. O For example, in 1968 some chemists studying the reactions that take place inside mass spectrometers needed to label the carbonyl oxygen atom of this ketone with the isotope 18 O. 16 18 By stirring the ‘normal’ O compound with a large excess of isotopically labelled water, H 2 O, for a few hours in the presence of a drop of acid they were able to make the required labelled com- í In Chapter 13 we saw this way of pound. Without the acid catalyst, the exchange is very slow. Acid catalysis speeds the reaction up by making a reaction go faster by raising making the carbonyl group more electrophilic so that equilibrium is reached more quickly. The the energy of the starting material. We 18 also saw that the position of an equilibrium is controlled by mass action— O is in large excess. -

Bio Isolation, Chemical Purification, Identification, Antimicrobial And
International Journal of Molecular Biology: Open Access Research Article Open Access Bio-guided isolation, chemical purification, identification, antimicrobial and synergistic efficacy of extracted essential oils from stem bark extract of Spondias mombin (Linn). Abstract Volume 4 Issue 4 - 2019 The purpose of this research work is to bio-guided isolate, purify, chemical identification, antimicrobial and Synergistic efficacy of extracted essential oils from ethyl acetate extract Oludare Temitope Osuntokun,1 Gamberini of Spondias mombin. The ethyl acetate stem bark extract of Spondias mombin were air- M Cristina2 dried, chopped into smaller pieces and cold extracted exhaustively with ethylacetate. The 1Department of Microbiology, Faculty of Science, Adekunle crude extract was partitioned using various solvents and the dichloromethane fraction was Ajasin University, Nigeria concentrated and fractionated using column chromatography packed with Silica gel and 2Department of Life Sciences, University of Modena and Reggio Sephadex-LH and eluted with appropriate solvent systems accordingly. In order to obtain Emilia, Italy pure extracts, partially purified fractions were further purified. The structures of the isolated compounds were determined by using data obtained from GC-MS spectrum. The compound Correspondence: Oludare Temitope Osuntokun, Department isolated includeAspidofractinine-3-methanol, Phthalic acid, 2-ethylhexyl tetradecyl of Microbiology, Faculty of Science, Adekunle Ajasin University, ester, Phthalic acid, di (2-propylpentyl) ester), (9-(2’, 2’-Dimethylpropanoilhydrazono)- Akungba-Akoko, Ondo state, Nigeria, 3,6-dichloro-2,7-bis-[2-(diethylamino)-ethoxy]fluorine) and Tere phthalic acid, dodecyl Email 2-ethylhexyl ester.These compounds have individual and synergistic activity against Gram Received: August 08, 2019 | Published: August 26, 2019 negative (E.coli), Gram-positive (Bacillus subtilis) and (Aspergillusflavus) at 10, 5, 2.5 and 1.25 g/mL. -

Correlation and Prediction of Mixing Thermodynamic Properties of Ester-Containing Systems: Ester+Alkane and Ester+Ester Binary S
J. Chem. Thermodynamics 54 (2012) 41–48 Contents lists available at SciVerse ScienceDirect J. Chem. Thermodynamics journal homepage: www.elsevier.com/locate/jct Correlation and prediction of mixing thermodynamic properties of ester-containing systems: Ester + alkane and ester + ester binary systems and the ternary dodecane + ethyl pentanoate + ethyl ethanoate ⇑ Noelia Pérez a, Luís Fernández a, Juan Ortega a, , Francisco J. Toledo a, Jaime Wisniak b a Laboratorio de Termodinámica y Fisicoquímica de Fluidos, Parque Científico-Tecnológico, Universidad de Las Palmas de Gran Canaria, Canary Islands, Spain b Deparment of Chemical Engineering, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel article info abstract Article history: E E Excess thermodynamic properties V m and Hm, have been measured for the ternary mixture dodecane + Received 25 December 2011 ethyl pentanoate + ethyl ethanoate and for the corresponding binaries dodecane + ethyl pentanoate, Received in revised form 7 March 2012 dodecane + ethyl ethanoate, ethyl pentanoate + ethyl ethanoate at 298.15 K. All mixtures show Accepted 9 March 2012 endothermic and expansive effects. Experimental results are correlated with a suitable equation whose Available online 20 March 2012 final form for the excess ternary quantity ME contains the particular contributions of the three binaries (i–j) and a last term corresponding to the ternary, all of them obtained considering fourth-order interac- Keywords: tions. The fit goodness for all mixtures is good and comparable to others equations taken from the liter- Excess molar enthalpies ature. In this work the dissolution model for the binaries and ternary is analyzed with a special attention Excess molar volumes Ternary mixture to ester–ester binaries whose behaviour is discussed. -

One-Step Synthesis of Ester from Alkene
ー論文ー ●One-step synthesis of ester from alkene Madoka Furuta M.C.Kung and H.H.Kung UFO Project Northwestern University Abstract A one-step synthesis of ethylacetate from a feed mixture of ethene, oxygen, and water using a supported Pd and silicotungstic acid catalyst was demonstrated. At about 180℃ and 25% ethene conversion, ethylacetate could be produced with to 46% selectivity, with 34% acetic acid and ethanol that could be recycled. The catalyst was believed to be bifunctional, with Pd providing the oxidation function and the silicotungstic acid providing the acidic function. And this catalyst system had an activity that could produce isopropylacrylate from a feed mixture of propene, oxygen, and water. 1 Introduction C2H5OH + O2 = CH3COOH + H2O (2) Carboxylic acid esters are used in a wide range of applications. They are used, for example, as plasticizers, solvents, surface-active Finally, ethyl acetate is produced by the esterification of ethanol agents, and flavor and perfume materials. There are many known with acetic acid over a catalyst. methods to produce esters. The most common method is the C2H5OH + CH3COOH = CH3COOC2H5 + H2O (3) esterification, which is a reaction of alcohol with carboxylic acid1,2). For example, ethyl acrylate is synthesized from ethyl alcohol and Thus the overall reaction is acrylic acid, and ethyl acetate from ethyl alcohol and acetic acid. An 2C2H4 + O2 = CH3COOC2H5 (4) acidic catalyst, which can be a mineral acid, or polymeric material containing chemically-bound acid groups, such as sulfonated Such a one-step process has the advantage of starting with reactants, polystyrene, is commonly used in the esterification. -
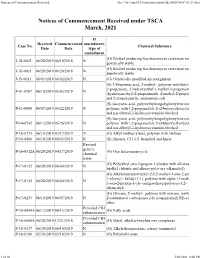
Notices of Commencement Received File:///W:/Oneepa/Newchems/Pubs/5D2-IMD/NOC-03-21.Html
Notices of Commencement Received file:///W:/OneEPA/Newchems/pubs/5d2-IMD/NOC-03-21.html If Received Commencement amendment, Case No. Chemical Substance Date Date type of amendment (G) Biofuel producing Saccharomyces cerevisiae mo J-18-0002 06/28/2019 06/19/2018 N genetically stable (G) Biofuel producing Saccharomyces cerevisiae mo J-18-0003 06/28/2019 06/24/2018 N genetically stable J-19-0011 06/03/2019 06/02/2019 N (G) Genetically modified microorganism (S) 2-Propenoic acid, 2-methyl-, polymer with butyl 2-propenoate, 2-hydroxyethyl 2-methyl-2-propenoat P-01-0767 06/10/2019 05/03/2019 N (hydroxymethyl)-2-propenamide, 2-methyl-2-propen and 2-propenenitrile, ammonium salt (S) Isocyanic acid, polymethylenepolyphenylene este P-11-0080 06/07/2019 05/22/2019 N polymer with 1,2-propanediol, 2-(2-butoxyethoxy)et and n,n-dibutyl-2-hydroxyacetamide-blocked (S) Isocyanic acid, polymethylenepolyphenylene este P-14-0742 06/13/2019 05/26/2019 N polymer with 1,2-propanediol, 2-(2-butoxyethoxy)et and n,n-dibutyl-2-hydroxyacetamide-blocked P-16-0375 06/10/2019 03/17/2019 N (G) Alkyl methacrylates, polymer with olefines P-16-0400 06/18/2019 06/03/2019 N (S) Alkanes, C11-16, branched and linear Revised generic P-16-0532A 06/20/2019 04/17/2019 (G) Oxo heteromonocycle chemical name (G) Poly(alkyl-oxo-2-propen-1-yl)ester with alkanea P-17-0152 06/28/2019 06/04/2019 N trialkyl chloride and alkoxy-poly(oxy-alkanediyl) (G) Alkylaminium-trialkyl-2-[(2-methyl-1-oxo-2-pro 1-yl)oxy]-, halide (1:1), polymer with alpha-(2-meth P-17-0152 06/28/2019 06/04/2019 N 1-oxo-2-propen-1-yl)- -

Educational Research Applications Abebe M, Et Al
Educational Research Applications Abebe M, et al. Educ Res Appl 5: 175. Review Article DOI: 10.29011/2575-7032.100175 Teaching Students Synthesizing Molecules Mimicking an Existing Drug against Covid-19 Moges Abebe1*, Lashan Eloise Knowles1, Bisrat Hailemeskel2 1Department of Biological and Physical Sciences, Saint Augustine University, Raleigh, NC, USA 2Department of Clinical & Administrative Pharmacy Sciences, College of Pharmacy, Howard University, NW Washington, DC, USA *Corresponding author: Moges Abebe, Department of Biological and Physical Sciences, Saint Augustine University, Raleigh, NC 27610, NC, USA Citation: Abebe M, Knowles LE, Hailemeskel B (2020) Teaching Students Synthesizing Molecules Mimicking an Existing Drug against Covid-19. Educ Res Appl 5: 175. DOI: 10.29011/2575-7032.100175 Received Date: 26 May 2020; Accepted Date: 01 June, 2020; Published Date: 06 June, 2020 Abstract End of semester organic chemistry course projects are valuable learning assessment tools while giving students a creative opportunity and sparking interest for further research investigations. The purpose of this year’s project was to teach students how to synthesize a molecule that potentially mimics an existing drug that works against the COVID-19. The available drugs chosen for the project are those that are proposed to work either by prohibiting the easy entry of the virus into respiratory tissues or those who deprive the virus’s ability to reproduce once they enter the cell. An investigative search in historical literature and the current conditions of the virus enabled students to create a unique and innovative product that requires a cumulative learned knowledge. History has shown that when a new virus becomes pandemic it takes time for researchers to create a drug, test the results, and gets approved by the Food and Drug Administration (FDA) for public availability. -

Ester Synthesis Lab (Student Handout)
Name: ________________________ Lab Partner: ____________________ Date: __________________________ Class Period: ____________________ Ester Synthesis Lab (Student Handout) Lab Report Components: The following must be included in your lab book in order to receive full credit. 1. Purpose 2. Hypothesis 3. Procedure 4. Observation/Data Table 5. Results 6. Mechanism (In class) 7. Conclusion Introduction The compounds you will be making are also naturally occurring compounds; the chemical structure of these compounds is already known from other investigations. Esters are organic molecules of the general form: where R1 and R2 are any carbon chain. Esters are unique in that they often have strong, pleasant odors. As such, they are often used in fragrances, and many artificial flavorings are in fact esters. Esters are produced by the reaction between alcohols and carboxylic acids. For example, reacting ethanol with acetic acid to give ethyl acetate is shown below. + → + In the case of ethyl acetate, R1 is a CH3 group and R2 is a CH3CH2 group. Naming esters systematically requires naming the functional groups on both sides of the bridging oxygen. In the example above, the right side of the ester as shown is a CH3CH2 1 group, or ethyl group. The left side is CH3C=O, or acetate. The name of the ester is therefore ethyl acetate. Deriving the names of the side from the carboxylic acid merely requires replacing the suffix –ic with –ate. Materials • Alcohol • Carboxylic Acid o 1 o A o 2 o B o 3 o C o 4 Observation Parameters: • Record the combination of carboxylic acid and alcohol • Observe each reactant • Observe each product Procedure 1. -

In This Handout, All of Our Functional Groups Are Presented As Condensed Line Formulas, 2D and 3D Formulas and with Nomenclature Prefixes and Suffixes (If Present)
In this handout, all of our functional groups are presented as condensed line formulas, 2D and 3D formulas and with nomenclature prefixes and suffixes (if present). Organic names are built on a foundation of alkanes, alkenes and alkynes. Those examples are presented first and you need to know those rules. The strategies can be found in Chapter 4 of our textbook (alkanes: pages 93-98, cycloalkanes 102-104, alkenes: pages 104-110, alkynes: pages 112-113 and combinations of all of them 113-115). After introducing examples of alkanes, alkenes, alkynes and combinations of them, the functional groups are presented in order of priority. A few nomenclature examples are provided for each of the functional groups. Examples of the various functional groups are presented on pages 115-135 in the textbook. Two overview pages are on pages 136-137. Some functional groups have a suffix name when they are the highest priority functional group and a prefix name when they are not the highest priority group, and these are added to the skeletal names with identifying numbers and stereochemistry terms (E and Z for alkenes, R and S for chiral centers and cis and trans for rings). Several low priority functional groups only have a prefix name. A few additional special patterns are shown on pages 98-102. The only way to learn this topic is practice (over and over). The best practice approach is to actually write out the names (on an extra piece of paper or on a white board, and then do it again). The same functional groups are used throughout the entire course.