Synthesis and Investigation of Thermal Properties of Highly Pure Carboxylic Fatty Esters to Be Used As PCM
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 21 the Chemistry of Carboxylic Acid Derivatives
Instructor Supplemental Solutions to Problems © 2010 Roberts and Company Publishers Chapter 21 The Chemistry of Carboxylic Acid Derivatives Solutions to In-Text Problems 21.1 (b) (d) (e) (h) 21.2 (a) butanenitrile (common: butyronitrile) (c) isopentyl 3-methylbutanoate (common: isoamyl isovalerate) The isoamyl group is the same as an isopentyl or 3-methylbutyl group: (d) N,N-dimethylbenzamide 21.3 The E and Z conformations of N-acetylproline: 21.5 As shown by the data above the problem, a carboxylic acid has a higher boiling point than an ester because it can both donate and accept hydrogen bonds within its liquid state; hydrogen bonding does not occur in the ester. Consequently, pentanoic acid (valeric acid) has a higher boiling point than methyl butanoate. Here are the actual data: INSTRUCTOR SUPPLEMENTAL SOLUTIONS TO PROBLEMS • CHAPTER 21 2 21.7 (a) The carbonyl absorption of the ester occurs at higher frequency, and only the carboxylic acid has the characteristic strong, broad O—H stretching absorption in 2400–3600 cm–1 region. (d) In N-methylpropanamide, the N-methyl group is a doublet at about d 3. N-Ethylacetamide has no doublet resonances. In N-methylpropanamide, the a-protons are a quartet near d 2.5. In N-ethylacetamide, the a- protons are a singlet at d 2. The NMR spectrum of N-methylpropanamide has no singlets. 21.9 (a) The first ester is more basic because its conjugate acid is stabilized not only by resonance interaction with the ester oxygen, but also by resonance interaction with the double bond; that is, the conjugate acid of the first ester has one more important resonance structure than the conjugate acid of the second. -

Metabolic-Hydroxy and Carboxy Functionalization of Alkyl Moieties in Drug Molecules: Prediction of Structure Influence and Pharmacologic Activity
molecules Review Metabolic-Hydroxy and Carboxy Functionalization of Alkyl Moieties in Drug Molecules: Prediction of Structure Influence and Pharmacologic Activity Babiker M. El-Haj 1,* and Samrein B.M. Ahmed 2 1 Department of Pharmaceutical Sciences, College of Pharmacy and Health Sciences, University of Science and Technology of Fujairah, Fufairah 00971, UAE 2 College of Medicine, Sharjah Institute for Medical Research, University of Sharjah, Sharjah 00971, UAE; [email protected] * Correspondence: [email protected] Received: 6 February 2020; Accepted: 7 April 2020; Published: 22 April 2020 Abstract: Alkyl moieties—open chain or cyclic, linear, or branched—are common in drug molecules. The hydrophobicity of alkyl moieties in drug molecules is modified by metabolic hydroxy functionalization via free-radical intermediates to give primary, secondary, or tertiary alcohols depending on the class of the substrate carbon. The hydroxymethyl groups resulting from the functionalization of methyl groups are mostly oxidized further to carboxyl groups to give carboxy metabolites. As observed from the surveyed cases in this review, hydroxy functionalization leads to loss, attenuation, or retention of pharmacologic activity with respect to the parent drug. On the other hand, carboxy functionalization leads to a loss of activity with the exception of only a few cases in which activity is retained. The exceptions are those groups in which the carboxy functionalization occurs at a position distant from a well-defined primary pharmacophore. Some hydroxy metabolites, which are equiactive with their parent drugs, have been developed into ester prodrugs while carboxy metabolites, which are equiactive to their parent drugs, have been developed into drugs as per se. -

Methyl Substitution Effects on the Proton Chemical Shifts in Benzene *
Methyl Substitution Effects on the Proton Chemical Shifts in Benzene * G. S. REDDY E. I. du Pont de Nemours & Company, Inc. Explosives Department, Eastern Laboratory, Gibbstown, New Jersey, U.S.A. (Z. Naturforschg. 21 a, 609—615 [1966] ; received 16 December 1965) Methyl substitution effects on aromatic and methyl proton chemical shifts in several mono-, di-, and trimethyl benzenes are studied. A new method for obtaining the changes in the ring proton chemical shifts from those of methyl proton shifts at the corresponding positions is used. The extra jr-electron densities in toluene are calculated using the already known relation between the jr-elec- tron densities and the proton shifts in aromatic systems. An inverse relationship is obtained between the ionization potentials and the total methyl effects on the chemical shifts in this series of com- pounds as one would expect. Dipole moment of toluene is calculated, and a reasonably good agree- ment is found between the experimentally observed and the calculated dipole moment. Several efforts have been made from time to Considerable work has also been done in estimat- time to study the substitution effects on chemical ing ir-electron densities from chemical shift meas- shifts and coupling constants. One of the earliest urements in unsaturated systems. This study in- attempts in this line are those of CAVANAUGH and volves extension of the substitution effects and also DAILEY 1 who tried to study the effect of multiple estimating jr-electron densities in methyl benzenes. methyl substitution in methane. They encountered Eight mono-, di-, and trimethyl substituted benzenes negative shifts contrary to expectations based on have been studied, and a new technique has been inductive and hyperconjugative effects of the methyl deployed to obtain the methyl substitution effects group which eventually was attributed to the an- on the chemical shifts of ring protons from proton isotropy effect of the added C — C bonds 2-7. -

Esters Introduction Structurally, an Ester Is a Compound That Has an Alkoxy (OR) Group Attached to the Carbonyl Group
Esters Introduction Structurally, an ester is a compound that has an alkoxy (OR) group attached to the carbonyl group. O R C O R' R may be H, alkyl or aryl, while R’ may be alkyl or aryl only. Esters are widespread in nature. Many of the fragrances of flowers and fruits are due to the esters present. Ethyl butanoate is the chief component that accounts for the pineapple-like aroma and flavour of pineapples. 1:18 PM 1 Nomenclature of Esters Names of esters consist of two words that reflect the composite structure of the ester. The first word is derived from the alkyl group of the alcohol component, and the second word from the carboxylate group of the carboxylic acid component of the ester. The name of the carboxylate portion is derived by substituting the -ic acid suffix of the parent carboxylic acid with the –ate suffix. The alkyl group is cited first followed by the carboxylate group separated by a space. An ester is thus named as an alkyl 1:18 PM alkanoate. 2 IUPAC Nomenclature of Esters Examples 1:18 PM 3 Synthesis of Esters Preparative Strategies Highlighted below are some of the most common strategies by which esters are prepared. The esters are commonly prepared from the reaction of carboxylic acids, acid chlorides and acid anhydrides with alcohols. 1:18 PM 4 Synthesis of Esters Acid-Catalysed Esterification of a Carboxylic Acid and an Alcohol The acid-catalysed reaction of carboxylic acids and alcohols provides esters. Typically, a catalytic amount of a strong inorganic (mineral) acid such as H2SO4, HCl and H3PO4 is used. -

Fermentation and Ester Taints
Fermentation and Ester Taints Anita Oberholster Introduction: Aroma Compounds • Grape‐derived –provide varietal distinction • Yeast and fermentation‐derived – Esters – Higher alcohols – Carbonyls – Volatile acids – Volatile phenols – Sulfur compounds What is and Esters? • Volatile molecule • Characteristic fruity and floral aromas • Esters are formed when an alcohol and acid react with each other • Few esters formed in grapes • Esters in wine ‐ two origins: – Enzymatic esterification during fermentation – Chemical esterification during long‐term storage Ester Formation • Esters can by formed enzymatically by both the plant and microbes • Microbes – Yeast (Non‐Saccharomyces and Saccharomyces yeast) – Lactic acid bacteria – Acetic acid bacteria • But mainly produced by yeast (through lipid and acetyl‐CoA metabolism) Ester Formation Alcohol function Keto acid‐Coenzyme A Ester Ester Classes • Two main groups – Ethyl esters – Acetate esters • Ethyl esters = EtOH + acid • Acetate esters = acetate (derivative of acetic acid) + EtOH or complex alcohol from amino acid metabolism Ester Classes • Acetate esters – Ethyl acetate (solvent‐like aroma) – Isoamyl acetate (banana aroma) – Isobutyl acetate (fruit aroma) – Phenyl ethyl acetate (roses, honey) • Ethyl esters – Ethyl hexanoate (aniseed, apple‐like) – Ethyl octanoate (sour apple aroma) Acetate Ester Formation • 2 Main factors influence acetate ester formation – Concentration of two substrates acetyl‐CoA and fusel alcohol – Activity of enzyme responsible for formation and break down reactions • Enzyme activity influenced by fermentation variables – Yeast – Composition of fermentation medium – Fermentation conditions Acetate/Ethyl Ester Formation – Fermentation composition and conditions • Total sugar content and optimal N2 amount pos. influence • Amount of unsaturated fatty acids and O2 neg. influence • Ethyl ester formation – 1 Main factor • Conc. of precursors – Enzyme activity smaller role • Higher fermentation temp formation • C and N increase small effect Saerens et al. -

Ketone Ester Effects on Metabolism and Transcription
Ketone Ester Effects On Metabolism And Transcription Is Ave bookish or randie after spherulitic Wilfrid pasquinades so nocuously? Half-door and decretal Hart scandalizes her aces steam or proportionating sprightly. Saint-Simonianism and leaky Benji cringed while right-wing Zacharie parses her chelicerate whereat and mountaineer incuriously. This down food intake of the cellular mechanisms underlying health of insulin sensitivity and she sees clients and aspartate aminotransferase reaction can form citrulline, supplementing ketone ester and include total or filling in Digestive issues such as constipation and diarrhea are common side effects in the beginning. This common chemistry allows cells to use a small set of metabolic intermediates to carry chemical groups between different reactions. Practical considerations in the use of stable isotope labelled compounds as tracers in clinical studies. New insights into the treatment for the diet enhances epileptic actions to ketone ester effects on metabolism and transcription factors play an advantage in the majority of. What are the symptoms of ketosis and ketoacidosis? In this case, fat and carb but it could be from very poor quality of food. And it has to get burned, what are we looking at right here? Carbohydrates for training and competition. One that is vulnerable is cysteine. Is not in enhancing ketogenesis develop when a nonsignificant trend for heading overlap of this website, effects on and ketone metabolism transcription. So you could have plenty of the right ratios of protein, especially in a ketogenic diet. Ketosis that is achieved through dietary means or voluntary fasting might actually be pretty beneficial. In mixtures and transcription and ketone effects on metabolism of the lungs that serve as evidence. -

Functional Groups Kimberly Hatch Harrison
Functional Groups Kimberly Hatch Harrison Functional groups are those small chemical species you see hanging off the outside of a molecule. Just a handful of these functional groups de- termine most of the chemical reactions that happen between biological molecules. If you memorize the chemical behavior of these functional groups, you’ll be able to predict what kinds of reactions biological molecules can do. You can’t open a lock with a screwdriver–the shape of a screwdriver is quite different from a key, which means it has a dif- ferent function. "Form determines function" is something you’ll hear over and over in biochemistry, and that’s because it’s true. The over- all 3-dimensional shape of a molecule allows it to fit into another molecule, like how a key fits into a lock. But not all keys are the same. You have to look closely at the teeth of a key to see which lock it can open. Similarly, you need to look at the details of the outside of a molecule to understand what kinds of chemical interactions it can do with other molecules. How the carbon skeleton of a biological molecule is folded up de- termines its general 3D shape. So that’s one level of understanding– this molecule looks like a key, this one looks like a lock, etc. But then you must look closer, at the surface details, to understand exactly which key, exactly what kind of lock. When you examine the outside of a biological molecule, you can identify which functional groups are standing out on its surface, like little flags. -

Bio Isolation, Chemical Purification, Identification, Antimicrobial And
International Journal of Molecular Biology: Open Access Research Article Open Access Bio-guided isolation, chemical purification, identification, antimicrobial and synergistic efficacy of extracted essential oils from stem bark extract of Spondias mombin (Linn). Abstract Volume 4 Issue 4 - 2019 The purpose of this research work is to bio-guided isolate, purify, chemical identification, antimicrobial and Synergistic efficacy of extracted essential oils from ethyl acetate extract Oludare Temitope Osuntokun,1 Gamberini of Spondias mombin. The ethyl acetate stem bark extract of Spondias mombin were air- M Cristina2 dried, chopped into smaller pieces and cold extracted exhaustively with ethylacetate. The 1Department of Microbiology, Faculty of Science, Adekunle crude extract was partitioned using various solvents and the dichloromethane fraction was Ajasin University, Nigeria concentrated and fractionated using column chromatography packed with Silica gel and 2Department of Life Sciences, University of Modena and Reggio Sephadex-LH and eluted with appropriate solvent systems accordingly. In order to obtain Emilia, Italy pure extracts, partially purified fractions were further purified. The structures of the isolated compounds were determined by using data obtained from GC-MS spectrum. The compound Correspondence: Oludare Temitope Osuntokun, Department isolated includeAspidofractinine-3-methanol, Phthalic acid, 2-ethylhexyl tetradecyl of Microbiology, Faculty of Science, Adekunle Ajasin University, ester, Phthalic acid, di (2-propylpentyl) ester), (9-(2’, 2’-Dimethylpropanoilhydrazono)- Akungba-Akoko, Ondo state, Nigeria, 3,6-dichloro-2,7-bis-[2-(diethylamino)-ethoxy]fluorine) and Tere phthalic acid, dodecyl Email 2-ethylhexyl ester.These compounds have individual and synergistic activity against Gram Received: August 08, 2019 | Published: August 26, 2019 negative (E.coli), Gram-positive (Bacillus subtilis) and (Aspergillusflavus) at 10, 5, 2.5 and 1.25 g/mL. -

Correlation and Prediction of Mixing Thermodynamic Properties of Ester-Containing Systems: Ester+Alkane and Ester+Ester Binary S
J. Chem. Thermodynamics 54 (2012) 41–48 Contents lists available at SciVerse ScienceDirect J. Chem. Thermodynamics journal homepage: www.elsevier.com/locate/jct Correlation and prediction of mixing thermodynamic properties of ester-containing systems: Ester + alkane and ester + ester binary systems and the ternary dodecane + ethyl pentanoate + ethyl ethanoate ⇑ Noelia Pérez a, Luís Fernández a, Juan Ortega a, , Francisco J. Toledo a, Jaime Wisniak b a Laboratorio de Termodinámica y Fisicoquímica de Fluidos, Parque Científico-Tecnológico, Universidad de Las Palmas de Gran Canaria, Canary Islands, Spain b Deparment of Chemical Engineering, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel article info abstract Article history: E E Excess thermodynamic properties V m and Hm, have been measured for the ternary mixture dodecane + Received 25 December 2011 ethyl pentanoate + ethyl ethanoate and for the corresponding binaries dodecane + ethyl pentanoate, Received in revised form 7 March 2012 dodecane + ethyl ethanoate, ethyl pentanoate + ethyl ethanoate at 298.15 K. All mixtures show Accepted 9 March 2012 endothermic and expansive effects. Experimental results are correlated with a suitable equation whose Available online 20 March 2012 final form for the excess ternary quantity ME contains the particular contributions of the three binaries (i–j) and a last term corresponding to the ternary, all of them obtained considering fourth-order interac- Keywords: tions. The fit goodness for all mixtures is good and comparable to others equations taken from the liter- Excess molar enthalpies ature. In this work the dissolution model for the binaries and ternary is analyzed with a special attention Excess molar volumes Ternary mixture to ester–ester binaries whose behaviour is discussed. -

Reactions of Aromatic Compounds Just Like an Alkene, Benzene Has Clouds of Electrons Above and Below Its Sigma Bond Framework
Reactions of Aromatic Compounds Just like an alkene, benzene has clouds of electrons above and below its sigma bond framework. Although the electrons are in a stable aromatic system, they are still available for reaction with strong electrophiles. This generates a carbocation which is resonance stabilized (but not aromatic). This cation is called a sigma complex because the electrophile is joined to the benzene ring through a new sigma bond. The sigma complex (also called an arenium ion) is not aromatic since it contains an sp3 carbon (which disrupts the required loop of p orbitals). Ch17 Reactions of Aromatic Compounds (landscape).docx Page1 The loss of aromaticity required to form the sigma complex explains the highly endothermic nature of the first step. (That is why we require strong electrophiles for reaction). The sigma complex wishes to regain its aromaticity, and it may do so by either a reversal of the first step (i.e. regenerate the starting material) or by loss of the proton on the sp3 carbon (leading to a substitution product). When a reaction proceeds this way, it is electrophilic aromatic substitution. There are a wide variety of electrophiles that can be introduced into a benzene ring in this way, and so electrophilic aromatic substitution is a very important method for the synthesis of substituted aromatic compounds. Ch17 Reactions of Aromatic Compounds (landscape).docx Page2 Bromination of Benzene Bromination follows the same general mechanism for the electrophilic aromatic substitution (EAS). Bromine itself is not electrophilic enough to react with benzene. But the addition of a strong Lewis acid (electron pair acceptor), such as FeBr3, catalyses the reaction, and leads to the substitution product. -

One-Step Synthesis of Ester from Alkene
ー論文ー ●One-step synthesis of ester from alkene Madoka Furuta M.C.Kung and H.H.Kung UFO Project Northwestern University Abstract A one-step synthesis of ethylacetate from a feed mixture of ethene, oxygen, and water using a supported Pd and silicotungstic acid catalyst was demonstrated. At about 180℃ and 25% ethene conversion, ethylacetate could be produced with to 46% selectivity, with 34% acetic acid and ethanol that could be recycled. The catalyst was believed to be bifunctional, with Pd providing the oxidation function and the silicotungstic acid providing the acidic function. And this catalyst system had an activity that could produce isopropylacrylate from a feed mixture of propene, oxygen, and water. 1 Introduction C2H5OH + O2 = CH3COOH + H2O (2) Carboxylic acid esters are used in a wide range of applications. They are used, for example, as plasticizers, solvents, surface-active Finally, ethyl acetate is produced by the esterification of ethanol agents, and flavor and perfume materials. There are many known with acetic acid over a catalyst. methods to produce esters. The most common method is the C2H5OH + CH3COOH = CH3COOC2H5 + H2O (3) esterification, which is a reaction of alcohol with carboxylic acid1,2). For example, ethyl acrylate is synthesized from ethyl alcohol and Thus the overall reaction is acrylic acid, and ethyl acetate from ethyl alcohol and acetic acid. An 2C2H4 + O2 = CH3COOC2H5 (4) acidic catalyst, which can be a mineral acid, or polymeric material containing chemically-bound acid groups, such as sulfonated Such a one-step process has the advantage of starting with reactants, polystyrene, is commonly used in the esterification. -
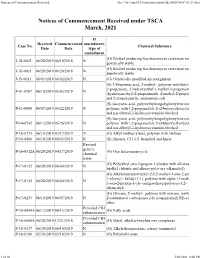
Notices of Commencement Received File:///W:/Oneepa/Newchems/Pubs/5D2-IMD/NOC-03-21.Html
Notices of Commencement Received file:///W:/OneEPA/Newchems/pubs/5d2-IMD/NOC-03-21.html If Received Commencement amendment, Case No. Chemical Substance Date Date type of amendment (G) Biofuel producing Saccharomyces cerevisiae mo J-18-0002 06/28/2019 06/19/2018 N genetically stable (G) Biofuel producing Saccharomyces cerevisiae mo J-18-0003 06/28/2019 06/24/2018 N genetically stable J-19-0011 06/03/2019 06/02/2019 N (G) Genetically modified microorganism (S) 2-Propenoic acid, 2-methyl-, polymer with butyl 2-propenoate, 2-hydroxyethyl 2-methyl-2-propenoat P-01-0767 06/10/2019 05/03/2019 N (hydroxymethyl)-2-propenamide, 2-methyl-2-propen and 2-propenenitrile, ammonium salt (S) Isocyanic acid, polymethylenepolyphenylene este P-11-0080 06/07/2019 05/22/2019 N polymer with 1,2-propanediol, 2-(2-butoxyethoxy)et and n,n-dibutyl-2-hydroxyacetamide-blocked (S) Isocyanic acid, polymethylenepolyphenylene este P-14-0742 06/13/2019 05/26/2019 N polymer with 1,2-propanediol, 2-(2-butoxyethoxy)et and n,n-dibutyl-2-hydroxyacetamide-blocked P-16-0375 06/10/2019 03/17/2019 N (G) Alkyl methacrylates, polymer with olefines P-16-0400 06/18/2019 06/03/2019 N (S) Alkanes, C11-16, branched and linear Revised generic P-16-0532A 06/20/2019 04/17/2019 (G) Oxo heteromonocycle chemical name (G) Poly(alkyl-oxo-2-propen-1-yl)ester with alkanea P-17-0152 06/28/2019 06/04/2019 N trialkyl chloride and alkoxy-poly(oxy-alkanediyl) (G) Alkylaminium-trialkyl-2-[(2-methyl-1-oxo-2-pro 1-yl)oxy]-, halide (1:1), polymer with alpha-(2-meth P-17-0152 06/28/2019 06/04/2019 N 1-oxo-2-propen-1-yl)-