Disorders of Acid-Base Balance 6.25
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Epileptic Seizure, As the First Symptom of Hypoparathyroidism in Children, Does Not Require Antiepileptic Drugs
Childs Nerv Syst DOI 10.1007/s00381-016-3264-2 ORIGINAL PAPER Epileptic seizure, as the first symptom of hypoparathyroidism in children, does not require antiepileptic drugs Meng-Jia Liu1 & Jiu-Wei Li2 & Xiu-Yu Shi1 & Lin-Yan Hu1 & Li-Ping Zou1,3 Received: 28 May 2016 /Accepted: 3 October 2016 # The Author(s) 2016. This article is published with open access at Springerlink.com Abstract Introduction Objective Patients with hypoparathyroidism exhibit metabol- ic disorders (hypocalcemia) and brain structural abnormalities Epileptic seizure occurs when a burst of electrical impulses in (brain calcifications). Currently, studies have determined the brain exceeds the normal limits. Its manifestation can vary whether antiepileptic drug (AED) treatment is required for from uncontrolled jerking movement (tonic–clonic seizure) to epileptic seizures in children with hypoparathyroidism. momentary loss of awareness (absence seizure). These im- Method This study aims to evaluate the data of two medical pulses spread to adjacent areas in the brain and create an un- centers in Beijing based on the diagnosis of epileptic seizures controlled storm of electrical activity. Brain diseases character- as the first symptom of hypoparathyroidism in children. ized by enduring predisposition to generate epileptic seizures Result A total of 42 patients were included and assigned into are collectively called epilepsy. According to pathogenesis, ep- AED and non-AED treatment groups in a 1:2 matched case– ilepsy can be classified into six categories: metabolic, structural, control study. Results show that the seizure outcome after inherited, immunologic, inflammatory, and idiopathic. 1 year of AED treatment is not significantly different from Hypoparathyroidism is an endocrine disease that results that of the control. -
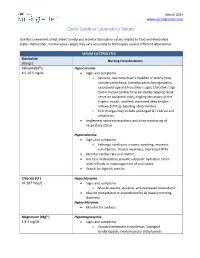
Quick Guide to Laboratory Values
March 2021 www.nursingcenter.com Quick Guide to Laboratory Values Use this convenient cheat-sheet to help you monitor laboratory values related to fluid and electrolyte status. Remember, normal value ranges may vary according to techniques used in different laboratories. SERUM ELECTROLYTES Electrolyte Nursing Considerations (Range) Calcium (Ca2+) Hypocalcemia 8.5-10.5 mg/dL • Signs and symptoms o Seizures, neuromuscular irritability or tetany (may include paresthesia, bronchospasm, laryngospasm, carpopedal spasm [Trousseau’s sign], Chvostek’s sign [facial muscle contractions elicited by tapping facial nerve on ipsilateral side], tingling sensations of the fingers, mouth, and feet, increased deep tendon reflexes [DTRs]), bleeding abnormalities o ECG changes may include prolonged QT interval and arrythmias. • Implement seizure precautions and close monitoring of respiratory status. Hypercalcemia • Signs and symptoms o Lethargy, confusion, nausea, vomiting, anorexia, constipation, muscle weakness, depressed DTRs • Monitor cardiac rate and rhythm. • Increase mobilization, provide adequate hydration either with IV fluids or encouragement of oral intake. • Watch for digitalis toxicity. Chloride (Cl-) Hypochloremia 97-107 mEq/L • Signs and symptoms o Muscle spasms, alkalosis, and depressed respirations • May be precipitated or exacerbated by GI losses (vomiting, diarrhea). Hyperchloremia • Monitor for acidosis. Magnesium (Mg2+) Hypomagnesemia 1.8-3 mg/dL • Signs and symptoms o Cardiac/ventricular arrhythmias, laryngeal stridor/spasm, neuromuscular -

Fluid & Electrolytes Fluid Balance Sodium 135-145 Meq/L
11/24/2009 Fluid & Electrolytes The Basics Fluid Balance Sodium 135‐145 meq/L • Imbalances typically associated with parallel changes in osmolality • Plays a major role in – ECF volume and concenttitration – Generation and transmission of nerve impulses – Acid–base balance 1 11/24/2009 Hypernatremia • Elevated serum sodium occurring with water loss or sodium gain • Causes hyperosmolality leading to cellular dehydration • Primary protection is thirst from hypothalamus Differential Assessment of ECF Volume Hypernatremia • Manifestations – Thirst, lethargy, agitation, seizures, and coma • Impaired LOC • Produced by clinical states – Central or nephrogenic diabetes insipidus – Serum sodium levels must be reduced gradually to avoid cerebral edema 2 11/24/2009 Nursing Management Nursing Diagnoses • Potential complication: seizures and coma leading to irreversible brain damage • Management • Treat undliderlying cause • If oral fluids cannot be ingested, IV solution of 5% dextrose in water or hypotonic saline • Diuretics Hyponatremia • Results from loss of sodium‐containing fluids or from water excess • Manifestations – CfiConfusion, nausea, vomiting, seizures, and coma Nursing Management Nursing Diagnoses • Risk for injury • Potential complication: severe neurologic changes • Management • Abnormal fluid loss – Fluid replacement with sodium‐containing solution • Caused by water excess – Fluid restriction is needed • Severe symptoms (seizures) – Give small amount of IV hypertonic saline solution (3% NaCl) 3 11/24/2009 Potassium 3.5‐5.5 meq/L • -

Hyperemesis Gravidarum with Paraparesis and Tetany
Open Access Case Report DOI: 10.7759/cureus.17014 Hyperemesis Gravidarum With Paraparesis and Tetany Jyotsnaa Muralitharan 1 , Vijayakumar Nagarajan 1 , Umarani Ravichandran 1 1. Internal Medicine, Rajah Muthiah Medical College & Hospital, Chidambaram, IND Corresponding author: Jyotsnaa Muralitharan, [email protected] Abstract Subacute-onset muscle weakness can result from channelopathies, inflammatory myopathies, thyroid dysfunction, hypoparathyroidism, vitamin D deficiency, and dyselectrolytemias like hypokalemia, hypocalcemia, and hypomagnesemia. We report a curious and extremely rare case of a 29-year-old woman with hyperemesis gravidarum presenting with disabling muscle weakness involving her lower limbs and trunk, and concurrent features of tetany. Following voluminous vomiting over the last two months, she presented with history of weakness of her lower limbs of 14 days duration, resulting in difficulty in her getting out of bed or walking unassisted. On examination, she was hypotensive (80/60 mmHg) and tachycardic (110 bpm), with flaccid weakness of her lower limbs (proximal weakness more than distal weakness - power of 1/5 at the hips bilaterally, and 3/5 at the knees and ankles bilaterally) and diminished deep tendon reflexes. She also had positive Trousseau’s sign and Chvostek’s sign. Interestingly, she also had thinned-out bluish sclerae, a high-arched palate, short stature, and bilateral conductive hearing loss. Laboratory evaluation revealed anemia, hyponatremia, hypokalemia, hypomagnesemia, hypochloremia, hypophosphatemia, and low vitamin D levels. Electrocardiogram showed prolonged QT interval. Her thyroid function test and parathyroid levels were normal. With parenteral replenishment of the electrolytes and vitamin D, her power improved and she was discharged on oral supplements. Thus, this case report demonstrates the importance of aggressive, early, and adequate management of hyperemesis gravidarum to prevent dyselectrolytemia-associated paraparesis. -

Calcium and Phosphorous Metabolism
CALCIUM AND PHOSPHOROUS METABOLISM -DR. SUHASINI. G P Lecturer Dept. of Oral Pathology & Microbiology Dr. Suhasini GP, Subharti Dental College, SVSU INTRODUCTION • Metabolism (Greek, Metabote= change) • Sum of the chemical and physical changes in tissue consisting of anabolism (reactions that convert small molecules into large) & catabolism (reactions that convert large molecules into small) Dr. Suhasini GP, Subharti Dental College, SVSU Functions of Calcium • Most abundant mineral • Most important inorganic cation in mineralised tissue- structural role • [Ca10(PO4)6(OH)2] in bone, dentin, cementum & enamel Dr. Suhasini GP, Subharti Dental College, SVSU Functions Formation of bone and teeth Controls secretion of glandular cells Muscle contraction Clotting of blood Neuromuscular excitability Cell-cell adhesion, cell integrity Intracellular second messenger in harmonal actions Dr. Suhasini GP, Subharti Dental College, SVSU Dietary requirements • Adults- 0.8 gms/day • Children & pregnant and lactating women- additional 1gm of Ca supplements/day • Postmenopausal women Dr. Suhasini GP, Subharti Dental College, SVSU Sources • Milk & its products • Green leafy veg (oxalic acid and phytic acid- reduces absorption) • Raagi • Egg yolk • Legumes • Nuts • Dried fish • Pan chewing with slacked lime • Drinking water Dr. Suhasini GP, Subharti Dental College, SVSU Distribution of Ca++` • Essentially distributed extracellularly • 1.2-1.4 kg in 70kg adult • 99%-skeleton • 1%- soft tissue+ body fluids Dr. Suhasini GP, Subharti Dental College, SVSU Bone Ca++ Exists in 2 forms- • Readily exchangeable form- simple exchange b/n newly deposited bone & exctracellular Ca++ • Stable form- constant formation & resorption of bone- – Controlled by PTH, calcitonin & vit.D Dr. Suhasini GP, Subharti Dental College, SVSU Blood Ca++ • Plasma, little in RBC • 9-11mg/dl- constant • In plasma- 1. -

Neurologic Complications of Electrolyte Disturbances and Acid–Base Balance
Handbook of Clinical Neurology, Vol. 119 (3rd series) Neurologic Aspects of Systemic Disease Part I Jose Biller and Jose M. Ferro, Editors © 2014 Elsevier B.V. All rights reserved Chapter 23 Neurologic complications of electrolyte disturbances and acid–base balance ALBERTO J. ESPAY* James J. and Joan A. Gardner Center for Parkinson’s Disease and Movement Disorders, Department of Neurology, UC Neuroscience Institute, University of Cincinnati, Cincinnati, OH, USA INTRODUCTION hyperglycemia or mannitol intake, when plasma osmolal- ity is high (hypertonic) due to the presence of either of The complex interplay between respiratory and renal these osmotically active substances (Weisberg, 1989; function is at the center of the electrolytic and acid-based Lippi and Aloe, 2010). True or hypotonic hyponatremia environment in which the central and peripheral nervous is always due to a relative excess of water compared to systems function. Neurological manifestations are sodium, and can occur in the setting of hypovolemia, accompaniments of all electrolytic and acid–base distur- euvolemia, and hypervolemia (Table 23.2), invariably bances once certain thresholds are reached (Riggs, reflecting an abnormal relationship between water and 2002). This chapter reviews the major changes resulting sodium, whereby the former is retained at a rate faster alterations in the plasma concentration of sodium, from than the latter (Milionis et al., 2002). Homeostatic mech- potassium, calcium, magnesium, and phosphorus as well anisms protecting against changes in volume and sodium as from acidemia and alkalemia (Table 23.1). concentration include sympathetic activity, the renin– angiotensin–aldosterone system, which cause resorption HYPONATREMIA of sodium by the kidneys, and the hypothalamic arginine vasopressin, also known as antidiuretic hormone (ADH), History and terminology which prompts resorption of water (Eiskjaer et al., 1991). -

Neurologic Complications of Electrolyte Disturbances and Acid–Base Balance
Handbook of Clinical Neurology, Vol. 119 (3rd series) Neurologic Aspects of Systemic Disease Part I Jose Biller and Jose M. Ferro, Editors © 2014 Elsevier B.V. All rights reserved Chapter 23 Neurologic complications of electrolyte disturbances and acid–base balance ALBERTO J. ESPAY* James J. and Joan A. Gardner Center for Parkinson’s Disease and Movement Disorders, Department of Neurology, UC Neuroscience Institute, University of Cincinnati, Cincinnati, OH, USA INTRODUCTION hyperglycemia or mannitol intake, when plasma osmolal- ity is high (hypertonic) due to the presence of either of The complex interplay between respiratory and renal these osmotically active substances (Weisberg, 1989; function is at the center of the electrolytic and acid-based Lippi and Aloe, 2010). True or hypotonic hyponatremia environment in which the central and peripheral nervous is always due to a relative excess of water compared to systems function. Neurological manifestations are sodium, and can occur in the setting of hypovolemia, accompaniments of all electrolytic and acid–base distur- euvolemia, and hypervolemia (Table 23.2), invariably bances once certain thresholds are reached (Riggs, reflecting an abnormal relationship between water and 2002). This chapter reviews the major changes resulting sodium, whereby the former is retained at a rate faster alterations in the plasma concentration of sodium, from than the latter (Milionis et al., 2002). Homeostatic mech- potassium, calcium, magnesium, and phosphorus as well anisms protecting against changes in volume and sodium as from acidemia and alkalemia (Table 23.1). concentration include sympathetic activity, the renin– angiotensin–aldosterone system, which cause resorption HYPONATREMIA of sodium by the kidneys, and the hypothalamic arginine vasopressin, also known as antidiuretic hormone (ADH), History and terminology which prompts resorption of water (Eiskjaer et al., 1991). -

Calcium Unresponsive Hypocalcemic Tetany: Gitelman Syndrome with Hypocalcemia
Hindawi Publishing Corporation Case Reports in Medicine Volume 2013, Article ID 197374, 3 pages http://dx.doi.org/10.1155/2013/197374 Case Report Calcium Unresponsive Hypocalcemic Tetany: Gitelman Syndrome with Hypocalcemia Madhav Desai, Praveen Kumar Kolla, and P. L. Venkata Pakki Reddy Department of Nephrology, Narayana Medical College, Chintareddypalem, Nellore, Andhra Pradesh 524003, India Correspondence should be addressed to Madhav Desai; [email protected] Received 4 May 2013; Accepted 20 August 2013 Academic Editor: Maxwell V. Meng Copyright © 2013 Madhav Desai et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Introduction. Gitelman’s syndrome (GS) is autosomal recessive renal tubular disorder characterized by hypokalemia, hypomag- nesemia, hypocalciuria, metabolic alkalosis, and hyperreninemic hyperaldosteronism. It is usually associated with normal serum calcium. We report a patient presented with hypocalcemic tetany, and evaluation showed Gitelman’s syndrome with hypocalcemia. Case Report.A28-year-oldwomanpresentedwithcrampsofthearms,legs,fatigue,andcarpalspasmsofoneweekduration.Shehas history of similar episodes on and off for the past two years. Her blood pressure was 98/66 mmHg. Chvostek’s sign and Trousseau’s sign were positive. Evaluation showed hypokalemia, hypocalcemia, hypomagnesemia, metabolic alkalosis, and hypocalciuria. Self-medication, diuretic use, laxative abuse, persistent vomiting, and diarrhoea were ruled out. Urinary prostaglandins and genetic testing could not be done because of nonavailability. To differentiate Gitelman syndrome from Bartter’s syndrome (BS), thiazide loading test was done. It showed blunted fractional chloride excretion. GS was confirmed and patient was treated with spironolactone along with magnesium, calcium, and potassium supplementation. -

Hypocalcemia: What a Surgeon Should Know 1Anish Kolly, 2Vijaya Sarathi, 3Sapana Bothra, 4Aromal Chekavar, 5Mayilvaganan Sabaretnam, 6Amit Agarwal
WJOES Anish Kolly et al 10.5005/jp-journals-10002-1215 HOW WE DO IT Hypocalcemia: What a Surgeon should know 1Anish Kolly, 2Vijaya Sarathi, 3Sapana Bothra, 4Aromal Chekavar, 5Mayilvaganan Sabaretnam, 6Amit Agarwal ABSTRACT of surgery, and differences in indication for surgery 5 Hypocalcemia is one of the sequelae following thyroidectomy across studies contribute to this wide variability. On the (TT) and becomes a complication when it becomes permanent. contrary, the risk of permanent postoperative hypocalce- Parathyroid preservation is a crucial step in the skillful opera- mia, which is defined as persistence of hypocalcemia even tive procedure of TT. When due care is not taken, the surgeon after 12 months of cervical surgery, has been reported to and the treating physician are faced with the issue of treating 6-9 the dreaded complication of permanent hypocalcemia. In this range from 0.6 to 12.1%. article, we address the issue of hypocalcemia following thyroid surgery and its management. ETIOPATHOGENESIS Keywords: Hypocalcemia, Parathyroid, Thyroidectomy. The cause of hypocalcemia after thyroid surgery is mul- How to cite this article: Kolly A, Sarathi V, Bothra S, tifactorial.10 Hypoparathyroidism seen after surgery can Chekavar A, Sabaretnam M, Agarwal A. Hypocalcemia: What be due to iatrogenic surgical trauma to the parathyroid a Surgeon should know. World J Endoc Surg 2017;9(2):72-77. glands or its blood supply or inadvertent removal of para- Source of support: Nil thyroid glands. Depending on the extent of parathyroid Conflict of interest: None gland damage, hypocalcemia may be transient, resolving within a few months, or permanent, in which the patient would require lifelong calcium and vitamin D supple- INTRODUCTION mentation. -

Hypomagnesemia with Hypocalcemia and Vitamin D Deficiency with Secondary Hypoparathyroidism
Case History Clinical Case Reports International Published: 22 Sep, 2020 Hypomagnesemia with Hypocalcemia and Vitamin D Deficiency with Secondary Hypoparathyroidism Bharat Parmar1* and Jwal Bharat2 1Department of Pediatrics, Zydus Medical College and Hospital, India 2Department of Pediatrics, Long Island University, USA Abstract The divalent cations, calcium and magnesium play vital role in neuromuscular function and cellular metabolism. Hypocalcemia related to parathyroid disorder, renal failure and bone disorders. The recent availability of ionized calcium measurement is helping to increase the accuracy of detection of this state. Evidence suggests that hypomagnesemia is a significant presence of refractory hypocalcemia, particularly in critical ill patient, with clinical significance relating mainly to concomitant electrolyte deficiency such hypokalemia. Case History A 10 year old child has came with generalized tonic-clonic convulsion first episode no history aura, loss of consciousness, bowel bladder incontinence was associated with up-rolling of eye balls, no froth from mouth no post seizure weakness or confusion, no history of precipitating factors dehydration, fever. Convulsion lasted for 4 min, normal birth history, and no similar episode in past, no history of convulsion in family, immunized up to age and normal development history. Past history of frequent spasms of hand peri-oral tingling, numbness. On clinical examination Chvostek’s sign and Trousseaus sign positive. Examination of central nervous system and endocrine was unremarkable, other system no abnormality, no abnormality in echocardiogram. Investigations Serum calcium 6.35 mg/dl, (8.4 mg/dl to 10.4 mg/dl), Ionized calcium 3.7 meq/l (4.3 meq/l to 5.3 meq/l), magnesium 1.2 mg/dl (1.7 mg/dl to 2.4 mg/dl), Serum alkaline phosphatase ALP 340 µ/L, Inorganic phosphorus 4.2 mg/dl (2.4 mg/dl to 4.5 mg/dl) serum albumin 3.75 gm/dl (3.2 gm/ OPEN ACCESS dl to 5 gm/dl). -

Fluid and Electrolyte Imbalances: Interpretation and Assessment
The Art and Science of Infusion Nursing Mandi D. Walker, MSN, RN-BC, CCRN Fluid and Electrolyte Imbalances: Interpretation and Assessment fluid. The other one-third is found in the extracellular ABSTRACT fluid and comprises water and sodium chloride. Maintaining the balance of fluid and electrolytes Extracellular fluid is further categorized as intravascular is crucial to the care of patients across the contin- and interstitial fluid. The body maintains a balance of uum. To do this, a practitioner must be cognizant fluid to molecules, including electrolytes, which is known 1-9 of key monitoring and assessment parameters. as homeostasis. Homeostasis is maintained through the transport of Key electrolytes, their function within the body, solutes through semipermeable membranes. This is normal values, signs and symptoms of imbalanc- accomplished using many different processes. The pro- es, key treatment modalities, and other considera- cess of diffusion occurs when the ions and molecules in tions are discussed. a solution spread out for uniform distribution. Filtration Key words: calcium, electrolyte imbalances, fluid is the process through which a liquid or gas passes balance, magnesium, phosphorus, potassium, through a filter (membrane) by forces of pressure. sodium Osmosis occurs when a solute passes through a mem- brane, leaving a solution of higher concentration to a solution of lower concentration, thereby balancing the n understanding of the normal balance of solutes on either side of the membrane. Finally, active fluid and electrolytes in the body is essen- transport is the process of solutes moving against the tial not only to understand normal func- electrochemical gradient, such as the sodium-potassium tioning of the human body but also to pump that moves the electrolytes back and forth predict problems and to intervene during depending on the need of the cell.1-10 Avarious disease processes. -

Tumor Lysis Syndrome
Oncologic Complications Resulting in Metabolic Disturbances: Tumor Lysis Syndrome Oncologic Complications Resulting in Metabolic Disturbances: Tumor Lysis Syndrome Authors: Belinda Mandrell, RN, MSN, St. Jude Children’s Research Hospital Ayda G. Nambayan, DSN, RN, St. Jude Children’s Research Hospital Content Reviewed by: Lisa South, RN, DSN, School of Nursing, University of Alabama at Birmingham Cure4Kids Release Date: 5 May 2006 Definition: Tumor lysis syndrome (A – 1) (TLS) is a metabolic disorder that results from the rapid release of intracellular contents (potassium, phosphorus and nucleic acids) into the circulation upon the rapid death of cells. The amounts of these intracellular products may exceed the body’s compensatory measures and cause four metabolic abnormalities: Hyperuricemia (A – 2) Hyperkalemia (A – 3) Hyperphosphatemia (A – 4) Hypocalcemia. Each of these electrolyte imbalances can occur individually or in combination, and severe increases in electrolyte concentrations can alter organ function. Risk Factors: Patients with large, rapidly growing tumors or pre-existing renal dysfunction are at greatest risk of TLS. Large tumor burdens may be clinically evident as hyperleukocytosis (increased white cell count [WBC] greater than 100,000/mm3), an increased concentration of lactose dehydrogenase (LDH), an increased concentration of uric acid, and massive enlargement of organs. Types of organ enlargement that might be seen are lymphadenopathy, a mediastinal mass and hepatosplenomegaly or other large abdominal tumors. Patients with chemosensitive tumors are also at risk of TLS. Therefore, this syndrome is most commonly associated with chemosensitive diseases that have a large tumor burden such as any leukemia with a high WBC count, Burkitt’s lymphoma and lymphocytic lymphoma.