Neurologic Complications of Electrolyte Disturbances and Acid–Base Balance
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Thursday 23 June 2016 – Morning A2 GCE HUMAN BIOLOGY F225/01 Genetics, Control and Ageing *5884237032* Candidates Answer on the Question Paper
Oxford Cambridge and RSA Thursday 23 June 2016 – Morning A2 GCE HUMAN BIOLOGY F225/01 Genetics, Control and Ageing *5884237032* Candidates answer on the Question Paper. OCR supplied materials: Duration: 2 hours None Other materials required: • Electronic calculator • Ruler (cm/mm) *F22501* INSTRUCTIONS TO CANDIDATES • Write your name, centre number and candidate number in the boxes above. Please write clearly and in capital letters. • Use black ink. HB pencil may be used for graphs and diagrams only. • Answer all the questions. • Read each question carefully. Make sure you know what you have to do before starting your answer. • Write your answer to each question in the space provided. If additional space is required, you should use the lined page(s) at the end of this booklet. The question number(s) must be clearly shown. • Do not write in the bar codes. INFORMATION FOR CANDIDATES • The number of marks is given in brackets [ ] at the end of each question or part question. • The total number of marks for this paper is 100. • Where you see this icon you will be awarded marks for the quality of written communication in your answer. • You may use an electronic calculator. • You are advised to show all the steps in any calculations. • This document consists of 24 pages. Any blank pages are indicated. © OCR 2016 [K/500/8502] OCR is an exempt Charity DC (NH/SW) 119808/4 Turn over 2 Answer all the questions. 1 Excretion is the removal of metabolic waste products from the body. The kidney is one of the organs involved in excretion. -

Prenatal Growth Restriction, Retinal Dystrophy, Diabetes Insipidus and White Matter Disease: Expanding the Spectrum of PRPS1-Related Disorders
European Journal of Human Genetics (2015) 23, 310–316 & 2015 Macmillan Publishers Limited All rights reserved 1018-4813/15 www.nature.com/ejhg ARTICLE Prenatal growth restriction, retinal dystrophy, diabetes insipidus and white matter disease: expanding the spectrum of PRPS1-related disorders Almundher Al-Maawali1,2, Lucie Dupuis1, Susan Blaser3, Elise Heon4, Mark Tarnopolsky5, Fathiya Al-Murshedi2, Christian R Marshall6,7, Tara Paton6,7, Stephen W Scherer6,7 for the FORGE Canada Consortium9, Jeroen Roelofsen8, Andre´ BP van Kuilenburg8 and Roberto Mendoza-Londono*,1 PRPS1 codes for the enzyme phosphoribosyl pyrophosphate synthetase-1 (PRS-1). The spectrum of PRPS1-related disorders associated with reduced activity includes Arts syndrome, Charcot–Marie–Tooth disease-5 (CMTX5) and X-linked non-syndromic sensorineural deafness (DFN2). We describe a novel phenotype associated with decreased PRS-1 function in two affected male siblings. Using whole exome and Sanger sequencing techniques, we identified a novel missense mutation in PRPS1. The clinical phenotype in our patients is characterized by high prenatal maternal a-fetoprotein, intrauterine growth restriction, dysmorphic facial features, severe intellectual disability and spastic quadraparesis. Additional phenotypic features include macular coloboma-like lesions with retinal dystrophy, severe short stature and diabetes insipidus. Exome sequencing of the two affected male siblings identified a shared putative pathogenic mutation c.586C4T p.(Arg196Trp) in the PRPS1 gene that was maternally inherited. Follow-up testing showed normal levels of hypoxanthine in urine samples and uric acid levels in blood serum. The PRS activity was significantly reduced in erythrocytes of the two patients. Nucleotide analysis in erythrocytes revealed abnormally low guanosine triphosphate and guanosine diphosphate. -

Epileptic Seizure, As the First Symptom of Hypoparathyroidism in Children, Does Not Require Antiepileptic Drugs
Childs Nerv Syst DOI 10.1007/s00381-016-3264-2 ORIGINAL PAPER Epileptic seizure, as the first symptom of hypoparathyroidism in children, does not require antiepileptic drugs Meng-Jia Liu1 & Jiu-Wei Li2 & Xiu-Yu Shi1 & Lin-Yan Hu1 & Li-Ping Zou1,3 Received: 28 May 2016 /Accepted: 3 October 2016 # The Author(s) 2016. This article is published with open access at Springerlink.com Abstract Introduction Objective Patients with hypoparathyroidism exhibit metabol- ic disorders (hypocalcemia) and brain structural abnormalities Epileptic seizure occurs when a burst of electrical impulses in (brain calcifications). Currently, studies have determined the brain exceeds the normal limits. Its manifestation can vary whether antiepileptic drug (AED) treatment is required for from uncontrolled jerking movement (tonic–clonic seizure) to epileptic seizures in children with hypoparathyroidism. momentary loss of awareness (absence seizure). These im- Method This study aims to evaluate the data of two medical pulses spread to adjacent areas in the brain and create an un- centers in Beijing based on the diagnosis of epileptic seizures controlled storm of electrical activity. Brain diseases character- as the first symptom of hypoparathyroidism in children. ized by enduring predisposition to generate epileptic seizures Result A total of 42 patients were included and assigned into are collectively called epilepsy. According to pathogenesis, ep- AED and non-AED treatment groups in a 1:2 matched case– ilepsy can be classified into six categories: metabolic, structural, control study. Results show that the seizure outcome after inherited, immunologic, inflammatory, and idiopathic. 1 year of AED treatment is not significantly different from Hypoparathyroidism is an endocrine disease that results that of the control. -

Acid-Base Abnormalities in Dogs with Diabetic Ketoacidosis: a Prospective Study of 60 Cases
325 Acid-base abnormalities in dogs with diabetic ketoacidosis: a prospective study of 60 cases Distúrbios ácido-base em cães com cetoacidose diabética: estudo prospectivo de 60 casos Ricardo DUARTE1; Denise Maria Nunes SIMÕES1; Khadine Kazue KANAYAMA1; Márcia Mery KOGIKA1 1School of Veterinary Medicine and Animal Science, University of São Paulo, São Paulo-SP, Brazil Abstract Diabetic ketoacidosis (DKA) is considered a typical high anion gap metabolic acidosis due to the retention of ketoanions. The objective of this study was to describe the acid-base disturbances of dogs with DKA and further characterize them, according to their frequency, adequacy of the secondary physiologic response, and occurrence of mixed disturbances. Sixty dogs with DKA were enrolled in the study. Arterial blood pH and gas tensions, plasma electrolytes, serum b-hydroxybutyrate (b-OHB), glucose, albumin and urea concentrations were determined for all dogs included in the study. All dogs were evaluated individually and systematically by the traditional approach to the diagnosis of acid- base disorders. Most of the dogs had a high anion gap acidosis, with appropriated respiratory response (n = 18; 30%) or concurrent respiratory alkalosis (n = 14; 23%). Hyperchloremic acidosis with moderated to marked increases in b-OHB was observed in 18 dogs (30%) and 7 of these patients had concurrent respiratory alkalosis. Hyperchloremic acidosis with mild increase in b-OHB was observed in 6 dogs (10%). Four dogs (7%) had a high anion gap acidosis with mild increase in b-OHB and respiratory alkalosis. Most of dogs with DKA had a high anion gap acidosis, but mixed acid-base disorders were common, chiefly high anion gap acidosis and concurrent respiratory alkalosis, and hyperchloremic acidosis with moderated to marked increases in serum b-OHB. -

Pathophysiology of Acid Base Balance: the Theory Practice Relationship
Intensive and Critical Care Nursing (2008) 24, 28—40 ORIGINAL ARTICLE Pathophysiology of acid base balance: The theory practice relationship Sharon L. Edwards ∗ Buckinghamshire Chilterns University College, Chalfont Campus, Newland Park, Gorelands Lane, Chalfont St. Giles, Buckinghamshire HP8 4AD, United Kingdom Accepted 13 May 2007 KEYWORDS Summary There are many disorders/diseases that lead to changes in acid base Acid base balance; balance. These conditions are not rare or uncommon in clinical practice, but every- Arterial blood gases; day occurrences on the ward or in critical care. Conditions such as asthma, chronic Acidosis; obstructive pulmonary disease (bronchitis or emphasaemia), diabetic ketoacidosis, Alkalosis renal disease or failure, any type of shock (sepsis, anaphylaxsis, neurogenic, cardio- genic, hypovolaemia), stress or anxiety which can lead to hyperventilation, and some drugs (sedatives, opoids) leading to reduced ventilation. In addition, some symptoms of disease can cause vomiting and diarrhoea, which effects acid base balance. It is imperative that critical care nurses are aware of changes that occur in relation to altered physiology, leading to an understanding of the changes in patients’ condition that are observed, and why the administration of some immediate therapies such as oxygen is imperative. © 2007 Elsevier Ltd. All rights reserved. Introduction the essential concepts of acid base physiology is necessary so that quick and correct diagnosis can The implications for practice with regards to be determined and appropriate treatment imple- acid base physiology are separated into respi- mented. ratory acidosis and alkalosis, metabolic acidosis The homeostatic imbalances of acid base are and alkalosis, observed in patients with differing examined as the body attempts to maintain pH bal- aetiologies. -
CURRENT Essentials of Nephrology & Hypertension
a LANGE medical book CURRENT ESSENTIALS: NEPHROLOGY & HYPERTENSION Edited by Edgar V. Lerma, MD Clinical Associate Professor of Medicine Section of Nephrology Department of Internal Medicine University of Illinois at Chicago College of Medicine Associates in Nephrology, SC Chicago, Illinois Jeffrey S. Berns, MD Professor of Medicine and Pediatrics Associate Dean for Graduate Medical Education The Perelman School of Medicine at the University of Pennsylvania Philadelphia, Pennsylvania Allen R. Nissenson, MD Emeritus Professor of Medicine David Geffen School of Medicine at UCLA Los Angeles, California Chief Medical Offi cer DaVita Inc. El Segundo, California New York Chicago San Francisco Lisbon London Madrid Mexico City Milan New Delhi San Juan Seoul Singapore Sydney Toronto Lerma_FM_p00i-xvi.indd i 4/27/12 10:33 AM Copyright © 2012 by The McGraw-Hill Companies, Inc. All rights reserved. Except as permitted under the United States Copyright Act of 1976, no part of this publication may be reproduced or distributed in any form or by any means, or stored in a database or retrieval system, without the prior written permission of the publisher. ISBN: 978-0-07-180858-3 MHID: 0-07-180858-2 The material in this eBook also appears in the print version of this title: ISBN: 978-0-07-144903-8, MHID: 0-07-144903-5. All trademarks are trademarks of their respective owners. Rather than put a trademark symbol after every occurrence of a trademarked name, we use names in an editorial fashion only, and to the benefi t of the trademark owner, with no intention of infringement of the trademark. -

Arterial Blood Gases: Acid-Base Balance
EDUCATIONAL COMMENTARY – ARTERIAL BLOOD GASES: ACID-BASE BALANCE Educational commentary is provided through our affiliation with the American Society for Clinical Pathology (ASCP). To obtain FREE CME/CMLE credits click on Earn CE Credits under Continuing Education on the left side of the screen. **Florida licensees, please note: This exercise will appear in CE Broker under the specialty of Blood Gas Analysis. LEARNING OUTCOMES On completion of this exercise, the participant should be able to • identify the important buffering systems in the human body. • explain the Henderson-Hasselbalch equation and its relationship to the bicarbonate/carbonic acid buffer system. • explain the different acid-base disorders, causes associated with them, and compensatory measures. • evaluate acid-base status using patient pH and pCO2 and bicarbonate levels. Introduction Arterial blood gas values are an important tool for assessing oxygenation and ventilation, evaluating acid- base status, and monitoring the effectiveness of therapy. The human body produces a daily net excess of acid through normal metabolic processes: cellular metabolism produces carbonic, sulfuric, and phosphoric acids. Under normal conditions, the body buffers accumulated hydrogen ions (H+) through a variety of buffering systems, the respiratory center, and kidneys to maintain a plasma pH of between 7.35 and 7.45. This tight maintenance of blood pH is essential: even slight changes in pH can alter the functioning of enzymes, the cellular uptake and use of metabolites, and the uptake and release of oxygen. Although diagnoses are made by physicians, laboratory professionals must be able to interpret arterial blood gas values to judge the validity of the laboratory results they report. -

1 Fluid and Elect. Disorders of Serum Sodium Concentration
DISORDERS OF SERUM SODIUM CONCENTRATION Bruce M. Tune, M.D. Stanford, California Regulation of Sodium and Water Excretion Sodium: glomerular filtration, aldosterone, atrial natriuretic factors, in response to the following stimuli. 1. Reabsorption: hypovolemia, decreased cardiac output, decreased renal blood flow. 2. Excretion: hypervolemia (Also caused by adrenal insufficiency, renal tubular disease, and diuretic drugs.) Water: antidiuretic honnone (serum osmolality, effective vascular volume), renal solute excretion. 1. Antidiuresis: hyperosmolality, hypovolemia, decreased cardiac output. 2. Diuresis: hypoosmolality, hypervolemia ~ natriuresis. Physiologic changes in renal salt and water excretion are more likely to favor conservation of normal vascular volume than nonnal osmolality, and may therefore lead to abnormalities of serum sodium concentration. Most commonly, 1. Hypovolemia -7 salt and water retention. 2. Hypervolemia -7 salt and water excretion. • HYFERNATREMIA Clinical Senini:: Sodium excess: salt-poisoning, hypertonic saline enemas Primary water deficit: chronic dehydration (as in diabetes insipidus) Mechanism: Dehydration ~ renal sodium retention, even during hypernatremia Rapid correction of hypernatremia can cause brain swelling - Management: Slow correction -- without rapid administration of free water (except in nephrogenic or untreated central diabetes insipidus) HYPONA1REMIAS Isosmolar A. Factitious: hyperlipidemia (lriglyceride-plus-plasma water volume). B. Other solutes: hyperglycemia, radiocontrast agents,. mannitol. -

Evaluation and Treatment of Alkalosis in Children
Review Article 51 Evaluation and Treatment of Alkalosis in Children Matjaž Kopač1 1 Division of Pediatrics, Department of Nephrology, University Address for correspondence Matjaž Kopač, MD, DSc, Division of Medical Centre Ljubljana, Ljubljana, Slovenia Pediatrics, Department of Nephrology, University Medical Centre Ljubljana, Bohoričeva 20, 1000 Ljubljana, Slovenia J Pediatr Intensive Care 2019;8:51–56. (e-mail: [email protected]). Abstract Alkalosisisadisorderofacid–base balance defined by elevated pH of the arterial blood. Metabolic alkalosis is characterized by primary elevation of the serum bicarbonate. Due to several mechanisms, it is often associated with hypochloremia and hypokalemia and can only persist in the presence of factors causing and maintaining alkalosis. Keywords Respiratory alkalosis is a consequence of dysfunction of respiratory system’s control ► alkalosis center. There are no pathognomonic symptoms. History is important in the evaluation ► children of alkalosis and usually reveals the cause. It is important to evaluate volemia during ► chloride physical examination. Treatment must be causal and prognosis depends on a cause. Introduction hydrogen ion concentration and an alkalosis is a pathologic Alkalosis is a disorder of acid–base balance defined by process that causes a decrease in the hydrogen ion concentra- elevated pH of the arterial blood. According to the origin, it tion. Therefore, acidemia and alkalemia indicate the pH can be metabolic or respiratory. Metabolic alkalosis is char- abnormality while acidosis and alkalosis indicate the patho- acterized by primary elevation of the serum bicarbonate that logic process that is taking place.3 can result from several mechanisms. It is the most common Regulation of hydrogen ion balance is basically similar to form of acid–base balance disorders. -

Cerebral Salt Wasting Syndrome and Systemic Lupus Erythematosus: Case Report
Elmer ress Case Report J Med Cases. 2016;7(9):399-402 Cerebral Salt Wasting Syndrome and Systemic Lupus Erythematosus: Case Report Filipe Martinsa, c, Carolina Ouriquea, Jose Faria da Costaa, Joao Nuakb, Vitor Braza, Edite Pereiraa, Antonio Sarmentob, Jorge Almeidaa Abstract disorders, that results in hyponatremia and a decrease in ex- tracellular fluid volume. It is characterized by a hypotonic hy- Cerebral salt wasting (CSW) is a rare cause of hypoosmolar hypona- ponatremia with inappropriately elevated urine sodium con- tremia usually associated with acute intracranial disease character- centration in the setting of a normal kidney function [1-3]. ized by extracellular volume depletion due to inappropriate sodium The onset of this disorder is typically seen within the first wasting in the urine. We report a case of a 46-year-old male with 10 days following a neurological insult and usually lasts no recently diagnosed systemic lupus erythematosus (SLE) initially pre- more than 1 week [1, 2]. Pathophysiology is not completely senting with neurological involvement and an antiphospholipid syn- understood but the major mechanism might be the inappropri- drome (APS) who was admitted because of chronic asymptomatic ate and excessive release of natriuretic peptides which would hyponatremia previously assumed as secondary to syndrome of inap- result in natriuresis and volume depletion. A secondary neu- propriate antidiuretic hormone secretion (SIADH). Initial evaluation rohormonal response would result in an increase in the renin- revealed a hypoosmolar hyponatremia with high urine osmolality angiotensin system and consequently in antidiuretic hormone and elevated urinary sodium concentration. Clinically, the patient’s (ADH) production. Since the volume stimulus is more potent extracellular volume status was difficult to define accurately. -

Managing Hyponatremia in Patients with Syndrome of Inappropriate Antidiuretic Hormone Secretion
REVIEW Managing Hyponatremia in Patients With Syndrome of Inappropriate Antidiuretic Hormone Secretion Joseph G. Verbalis, MD Division of Endocrinology and Metabolism, Department of Medicine, Georgetown University Medical Center, Washington DC. J.G. Verbalis received an honorarium funded by an unrestricted educational grant from Otsuka America Pharmaceuticals, Inc., for time and expertise spent in the composition of this article. No editorial assistance was provided. No other conflicts exist. This review will address the management of hyponatremia caused by the syndrome of inappropriate antidiuretic hormone secretion (SIADH) in hospitalized patients. To do so requires an understanding of the pathogenesis and diagnosis of SIADH, as well as currently available treatment options. The review will be structured as responses to a series of questions, followed by a presentation of an algorithm for determining the most appropriate treatments for individual patients with SIADH based on their presenting symptoms. Journal of Hospital Medicine 2010;5:S18–S26. VC 2010 Society of Hospital Medicine. Why is SIADH Important to Hospitalists? What Causes Hyponatremia in Patients with SIADH? Disorders of body fluids, and particularly hyponatremia, are Hyponatremia can be caused by 1 of 2 potential disruptions among the most commonly encountered problems in clinical in fluid balance: dilution from retained water, or depletion medicine, affecting up to 30% of hospitalized patients. In a from electrolyte losses in excess of water. Dilutional hypo- study of 303,577 laboratory samples collected from 120,137 natremias are associated with either a normal (euvolemic) patients, the prevalence of hyponatremia (serum [Naþ] <135 or an increased (hypervolemic) extracellular fluid (ECF) vol- mmol/L) on initial presentation to a healthcare provider was ume, whereas depletional hyponatremias generally are asso- 28.2% among those treated in an acute hospital care setting, ciated with a decreased ECF volume (hypovolemic). -
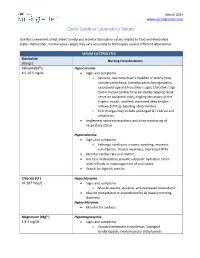
Quick Guide to Laboratory Values
March 2021 www.nursingcenter.com Quick Guide to Laboratory Values Use this convenient cheat-sheet to help you monitor laboratory values related to fluid and electrolyte status. Remember, normal value ranges may vary according to techniques used in different laboratories. SERUM ELECTROLYTES Electrolyte Nursing Considerations (Range) Calcium (Ca2+) Hypocalcemia 8.5-10.5 mg/dL • Signs and symptoms o Seizures, neuromuscular irritability or tetany (may include paresthesia, bronchospasm, laryngospasm, carpopedal spasm [Trousseau’s sign], Chvostek’s sign [facial muscle contractions elicited by tapping facial nerve on ipsilateral side], tingling sensations of the fingers, mouth, and feet, increased deep tendon reflexes [DTRs]), bleeding abnormalities o ECG changes may include prolonged QT interval and arrythmias. • Implement seizure precautions and close monitoring of respiratory status. Hypercalcemia • Signs and symptoms o Lethargy, confusion, nausea, vomiting, anorexia, constipation, muscle weakness, depressed DTRs • Monitor cardiac rate and rhythm. • Increase mobilization, provide adequate hydration either with IV fluids or encouragement of oral intake. • Watch for digitalis toxicity. Chloride (Cl-) Hypochloremia 97-107 mEq/L • Signs and symptoms o Muscle spasms, alkalosis, and depressed respirations • May be precipitated or exacerbated by GI losses (vomiting, diarrhea). Hyperchloremia • Monitor for acidosis. Magnesium (Mg2+) Hypomagnesemia 1.8-3 mg/dL • Signs and symptoms o Cardiac/ventricular arrhythmias, laryngeal stridor/spasm, neuromuscular