Quinones Are Growth Factors for the Human Gut Microbiota
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Goble Biochemistry 2
Subscriber access provided by - Access paid by the | UCSF Library Article Deamination of 6-Aminodeoxyfutalosine in Menaquinone Biosynthesis by Distantly Related Enzymes Alissa Marie Goble, Rafael Toro, Xu Li, Argentina Ornelas, Hao Fan, Subramaniam Eswaramoorthy, Yury V. Patskovsky, Brandan Hillerich, Ronald D. Seidel, Andrej Sali, Brian K. Shoichet, Steven C. Almo, Subramanyam Swaminathan, Martin E. Tanner, and Frank Michael Raushel Biochemistry, Just Accepted Manuscript • DOI: 10.1021/bi400750a • Publication Date (Web): 23 Aug 2013 Downloaded from http://pubs.acs.org on August 28, 2013 Just Accepted “Just Accepted” manuscripts have been peer-reviewed and accepted for publication. They are posted online prior to technical editing, formatting for publication and author proofing. The American Chemical Society provides “Just Accepted” as a free service to the research community to expedite the dissemination of scientific material as soon as possible after acceptance. “Just Accepted” manuscripts appear in full in PDF format accompanied by an HTML abstract. “Just Accepted” manuscripts have been fully peer reviewed, but should not be considered the official version of record. They are accessible to all readers and citable by the Digital Object Identifier (DOI®). “Just Accepted” is an optional service offered to authors. Therefore, the “Just Accepted” Web site may not include all articles that will be published in the journal. After a manuscript is technically edited and formatted, it will be removed from the “Just Accepted” Web site and published as an ASAP article. Note that technical editing may introduce minor changes to the manuscript text and/or graphics which could affect content, and all legal disclaimers and ethical guidelines that apply to the journal pertain. -

All Enzymes in BRENDA™ the Comprehensive Enzyme Information System
All enzymes in BRENDA™ The Comprehensive Enzyme Information System http://www.brenda-enzymes.org/index.php4?page=information/all_enzymes.php4 1.1.1.1 alcohol dehydrogenase 1.1.1.B1 D-arabitol-phosphate dehydrogenase 1.1.1.2 alcohol dehydrogenase (NADP+) 1.1.1.B3 (S)-specific secondary alcohol dehydrogenase 1.1.1.3 homoserine dehydrogenase 1.1.1.B4 (R)-specific secondary alcohol dehydrogenase 1.1.1.4 (R,R)-butanediol dehydrogenase 1.1.1.5 acetoin dehydrogenase 1.1.1.B5 NADP-retinol dehydrogenase 1.1.1.6 glycerol dehydrogenase 1.1.1.7 propanediol-phosphate dehydrogenase 1.1.1.8 glycerol-3-phosphate dehydrogenase (NAD+) 1.1.1.9 D-xylulose reductase 1.1.1.10 L-xylulose reductase 1.1.1.11 D-arabinitol 4-dehydrogenase 1.1.1.12 L-arabinitol 4-dehydrogenase 1.1.1.13 L-arabinitol 2-dehydrogenase 1.1.1.14 L-iditol 2-dehydrogenase 1.1.1.15 D-iditol 2-dehydrogenase 1.1.1.16 galactitol 2-dehydrogenase 1.1.1.17 mannitol-1-phosphate 5-dehydrogenase 1.1.1.18 inositol 2-dehydrogenase 1.1.1.19 glucuronate reductase 1.1.1.20 glucuronolactone reductase 1.1.1.21 aldehyde reductase 1.1.1.22 UDP-glucose 6-dehydrogenase 1.1.1.23 histidinol dehydrogenase 1.1.1.24 quinate dehydrogenase 1.1.1.25 shikimate dehydrogenase 1.1.1.26 glyoxylate reductase 1.1.1.27 L-lactate dehydrogenase 1.1.1.28 D-lactate dehydrogenase 1.1.1.29 glycerate dehydrogenase 1.1.1.30 3-hydroxybutyrate dehydrogenase 1.1.1.31 3-hydroxyisobutyrate dehydrogenase 1.1.1.32 mevaldate reductase 1.1.1.33 mevaldate reductase (NADPH) 1.1.1.34 hydroxymethylglutaryl-CoA reductase (NADPH) 1.1.1.35 3-hydroxyacyl-CoA -

Advances in Enhanced Menaquinone-7 Production from Bacillus Subtilis
REVIEW published: 19 July 2021 doi: 10.3389/fbioe.2021.695526 Advances in Enhanced Menaquinone-7 Production From Bacillus subtilis Chaoyong Liao 1, Hammed Ayansola 1, Yanbo Ma 2, Koichi Ito 3, Yuming Guo 1 and Bingkun Zhang 1* 1State Key Laboratory of Animal Nutrition, Department of Animal Nutrition and Feed Science, College of Animal Science and Technology, China Agricultural University, Beijing, China, 2Henan International Joint Laboratory of Animal Welfare and Health Breeding, Department of Animal Physiology, College of Animal Science and Technology, Henan University of Science and Technology, Luoyang, China, 3Department of Food and Physiological Models, Graduate School of Agricultural and Life Sciences, The University of Tokyo, Ibaraki, Japan The production of nutraceutical compounds through biosynthetic approaches has received considerable attention in recent years. For example, Menaquinone-7 (MK-7), a sub-type of Vitamin K2, biosynthesized from Bacillus subtilis (B. subtilis), proved to be more efficiently produced than the conventional chemical synthesis techniques. This is possible due to the development of B. subtilis as a chassis cell during the biosynthesis stages. Hence, it is imperative to provide insights on the B. subtilis membrane permeability modifications, biofilm reactors, and fermentation optimization as advanced techniques relevant to MK-7 production. Although the traditional gene-editing method of homologous Edited by: recombination improves the biosynthetic pathway, CRISPR-Cas9 could potentially resolve Tao Chen, Tianjin University, China the drawbacks of traditional genome editing techniques. For these reasons, future studies Reviewed by: should explore the applications of CRISPRi (CRISPR interference) and CRISPRa (CRISPR Yanfeng Liu, activation) system gene-editing tools in the MK-7 anabolism pathway. -

Engineering Elizabethkingia Meningoseptica Sp. F2 for Vitamin K2 Production Guided by Genome Analysis
Engineering Elizabethkingia meningoseptica sp. F2 for Vitamin K2 production guided by genome analysis. Qiang Yang Chinese Acadamy of science zhiming zheng ( [email protected] ) Hefei Institutes of Physical Science, Chinese Academy of Sciences https://orcid.org/0000-0002-0550- 8087 Hui Liu Chinese Acadamy science Peng Wang Chinese Acadamy of science Li Wang Chinese Acadamy science Xiaowen Sun Chinse Acadamy of science Wenfeng Ni Chinese Acadamy of science Han Wang Chinese Acadamy of Science Hengfang Tang Chinese Acadamy of science Genhai Zhao Chinese Acadamy of science Research Keywords: Vitamin K2, Elizabethkingia meningoseptica, Comparative genomics, metabolic network analysis Posted Date: May 1st, 2020 DOI: https://doi.org/10.21203/rs.3.rs-25351/v1 Page 1/24 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 2/24 Abstract Background The species in family Elizabethkingia meningoseptica are interesting strain for investigating Vitamin K2 metabolic analysis. However, their genomic sequence, metabolic pathway, potential abilities, and evolutionary status are still unknown. Results This study therefore aimed to perform a genome sequencing of Elizabethkingia meningoseptica sp. F2 and further accomplished comparative analysis with other Vitamin K2 strains reveals overall identifying its unique/shared metabolic genes across genomes. The 3,874,794–base pair sequence of Elizabethkingia meningoseptica sp. F2 is presented. Of 3,539 genes annotation was applied. Results of synteny block demonstrated Elizabethkingia meningoseptica sp. F2 shares high levels of synteny with Elizabethkingia meningoseptica ATCC 13253 and Elizabethkingia meningoseptica NBRC 12535. Identication of Vitamin K2 metabolic pathway in Elizabethkingia meningoseptica sp. F2 were also accomplished. -

Mechanisms for Extracellular Electron Exchange by Geobacter Species
University of Massachusetts Amherst ScholarWorks@UMass Amherst Doctoral Dissertations Dissertations and Theses Spring March 2015 Mechanisms for Extracellular Electron Exchange by Geobacter Species Jessica A. Smith University of Massachusetts Amherst Follow this and additional works at: https://scholarworks.umass.edu/dissertations_2 Part of the Environmental Microbiology and Microbial Ecology Commons, and the Microbial Physiology Commons Recommended Citation Smith, Jessica A., "Mechanisms for Extracellular Electron Exchange by Geobacter Species" (2015). Doctoral Dissertations. 325. https://doi.org/10.7275/6457832.0 https://scholarworks.umass.edu/dissertations_2/325 This Open Access Dissertation is brought to you for free and open access by the Dissertations and Theses at ScholarWorks@UMass Amherst. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of ScholarWorks@UMass Amherst. For more information, please contact [email protected]. MECHANISMS FOR EXTRACELLULAR ELECTRON EXCHANGE BY GEOBACTER SPECIES A Dissertation Presented by JESSICA AMBER SMITH Submitted to the Graduate School of the University of Massachusetts Amherst in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY February 2015 Department of Microbiology © Copyright by Jessica Amber Smith 2015 All Rights Reserved MECHANISMS FOR EXTRACELLULAR ELECTRON EXCHANGE BY GEOBACTER SPECIES A Dissertation Presented by JESSICA AMBER SMITH Approved as to style and content by: ________________________________ Derek R. Lovley, Chair ________________________________ James F. Holden, Member ________________________________ Steven J. Sandler, Member ________________________________ Dawn E. Holmes, Member ____________________________ John M. Lopes, Department Head Department of Microbiology ACKNOWLEDGMENTS I would like to thank my advisor, Derek Lovley, and the members of my committee, James Holden, Steven Sandler, and Dawn Holmes, for supporting and guiding my projects. -

Deamination of 6‑Aminodeoxyfutalosine in Menaquinone Biosynthesis by Distantly Related Enzymes † ∥ # † ‡ § ⊥ ¶ Alissa M
Article pubs.acs.org/biochemistry Deamination of 6‑Aminodeoxyfutalosine in Menaquinone Biosynthesis by Distantly Related Enzymes † ∥ # † ‡ § ⊥ ¶ Alissa M. Goble, Rafael Toro, Xu Li, Argentina Ornelas, Hao Fan, , , Subramaniam Eswaramoorthy, ∥ ∥ ∥ ‡ ⊥ § ⊥ ∥ Yury Patskovsky, Brandan Hillerich, Ron Seidel, Andrej Sali, , Brian K. Shoichet, , Steven C. Almo, ¶ # † Subramanyam Swaminathan, Martin E. Tanner, and Frank M. Raushel*, † Department of Chemistry, Texas A&M University, P.O. Box 30012, College Station, Texas 77843-3012, United States ‡ § ⊥ Department of Bioengineering and Theraputic Sciences, Department of Pharmaceutical Chemistry, and California Institute for Quantitative Biosciences, University of CaliforniaSan Francisco, 1700 Fourth Street, San Francisco, California 94158, United States ¶ Biology Department, Brookhaven National Laboratory, P.O. Box 5000, Upton, New York 11973-5000, United States # Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, British Columbia V6T 1Z1, Canada ∥ Department of Biochemistry, Einstein College of Medicine, Bronx, New York 10461, United States ABSTRACT: Proteins of unknown function belonging to cog1816 and cog0402 were characterized. Sav2595 from Steptomyces avermitilis MA-4680, Acel0264 from Acidothermus cellulolyticus 11B, Nis0429 from Nitratiruptor sp. SB155-2 and Dr0824 from Deinococcus radiodurans R1 were cloned, purified, and their substrate profiles determined. These enzymes were previously incorrectly annotated as adenosine deaminases or chlorohydrolases. It was shown here that these enzymes actually deaminate 6-aminodeoxyfutalosine. The deamination of 6-aminodeoxyfutalosine is part of an alternative menaquinone biosynthetic pathway that involves the formation of futalosine. 6-Aminodeoxyfutalosine is deaminated by these enzymes with ffi 5 −1 −1 − μ − −1 ′ catalytic e ciencies greater than 10 M s , Km values of 0.9 6.0 M, and kcat values of 1.2 8.6 s . -

(19) United States (12) Patent Application Publication (10) Pub
US 20130244920A1 (19) United States (12) Patent Application Publication (10) Pub. N0.: US 2013/0244920 A1 Lee et al. (43) Pub. Date: Sep. 19, 2013 (54) WATER SOLUBLE COMPOSITIONS (52) US. Cl. INCORPORATING ENZYMES, AND METHOD USPC ......................................... .. 510/392; 264/299 OF MAKING SAME (57) ABSTRACT (76) Inventors: David M. Lee, CroWn Point, IN (US); Jennifer L‘ Sims’ Lowell’ IN (Us) Disclosed herein are Water soluble compositions, such as ?lms, including a mixture of a ?rst Water-soluble resin, an (21) Appl' NO': 13/422’709 enzyme, and an enzyme stabilizer Which comprises a func (22) Filed: Man 16, 2012 tional substrate for the enzyme, methods of making such compositions, and methods of using such compositions, e.g. Publication Classi?cation to make packets containing functional ingredients. The enzymes can include proteases and mixtures of proteases (51) Int. Cl. With other enzymes, and the compositions provide good C11D 3/386 (2006.01) retention of enzyme function following ?lm processing and B29C 39/02 (2006.01) storage. US 2013/0244920 A1 Sep. 19,2013 WATER SOLUBLE COMPOSITIONS preheated to a temperature less than 77° C., optionally in a INCORPORATING ENZYMES, AND METHOD range ofabout 66° C. to about 77° C., or about 74° C.; drying OF MAKING SAME the Water from the cast mixture over a period of less than 24 hours, optionally less than 12 hours, optionally less than 8 FIELD OF THE DISCLOSURE hours, optionally less than 2 hours, optionally less than 1 [0001] The present disclosure relates generally to Water hour, optionally less than 45 minutes, optionally less than 30 soluble ?lms. -
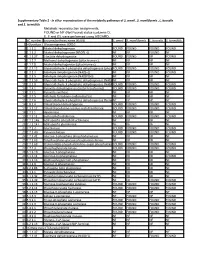
Supplementary Table 2 - in Silico Reconstruction of the Metabolic Pathways of S
Supplementary Table 2 - In silico reconstruction of the metabolic pathways of S. amnii , S. moniliformis , L. buccalis and S. termiditis Metabolic reconstruction assignments, FOUND or NF (Not Found) status (columns D, E, F and G), were performed using ASGARD, EC number Enzyme/pathway name (KEGG) S. amnii S. moniliformis L. buccalis S. termiditis 1 >Glycolysis / Gluconeogenesis 00010 2 1.1.1.1 Alcohol dehydrogenase. FOUND FOUND FOUND FOUND 3 1.1.1.2 Alcohol dehydrogenase (NADP(+)). NF NF FOUND NF 4 1.1.1.27 L-lactate dehydrogenase. FOUND FOUND NF FOUND 5 1.1.2.7 Methanol dehydrogenase (cytochrome c). NF NF NF NF 6 1.1.2.8 Alcohol dehydrogenase (cytochrome c). NF NF NF NF 7 1.2.1.12 Glyceraldehyde-3-phosphate dehydrogenase (phosphorylating).FOUND FOUND FOUND FOUND 8 1.2.1.3 Aldehyde dehydrogenase (NAD(+)). NF NF FOUND FOUND 9 1.2.1.5 Aldehyde dehydrogenase (NAD(P)(+)). NF NF NF NF 10 1.2.1.59 Glyceraldehyde-3-phosphate dehydrogenase (NAD(P)(+))NF (phosphorylating).NF NF NF 11 1.2.1.9 Glyceraldehyde-3-phosphate dehydrogenase (NADP(+)).FOUND FOUND FOUND FOUND 12 1.2.4.1 Pyruvate dehydrogenase (acetyl-transferring). FOUND FOUND FOUND FOUND 13 1.2.7.1 Pyruvate synthase. NF NF NF NF 14 1.2.7.5 Aldehyde ferredoxin oxidoreductase. NF NF NF NF 15 1.2.7.6 Glyceraldehyde-3-phosphate dehydrogenase (ferredoxin).NF NF NF NF 16 1.8.1.4 Dihydrolipoyl dehydrogenase. FOUND FOUND FOUND FOUND 17 2.3.1.12 Dihydrolipoyllysine-residue acetyltransferase. FOUND FOUND FOUND FOUND 18 2.7.1.1 Hexokinase. -

Molecular Investigation of Australian Termites and Their Gut Symbionts
Molecular Investigation of Australian Termites and their Gut Symbionts Nurdyana Abdul Rahman BBiotech (Hons), University of Queensland A thesis submitted for the degree of Doctor of Philosophy at The University of Queensland in 2015 School of Chemistry and Molecular Biosciences Abstract Termites are one of the most abundant and ecologically important eusocial insects in tropical and subtropical regions. Their success as lignocellulose decomposers is a result of a mutualistic relationship with their gut microbiota. Termites have evolved from wood-feeding cockroaches (lower termites) and expanded their dietary scope to soil, herbivore dung, grass and litter (higher termites). The introduction of high-throughput culture-independent molecular techniques has reinvigorated efforts to understand the termite gut microbiome and its involvement in symbiotic digestion. Yet, there remain significant unanswered or poorly answered questions regarding termite gut microbiome ecology and evolution such as the relative effect of diet vs vertical inheritance on shaping gut communities, the resilience of these communities under changing dietary regimes, the function of specific populations and the relative contributions of prokaryotic and eukaryotic symbionts to hydrolysis in lower termites. The aim of this thesis is to address these questions making use of Australia’s diverse but understudied termite species. In Chapter 2, a molecular survey using SSU rRNA amplicon pyrosequencing was conducted on 66 termite gut samples comprising seven higher termite genera and nine lower termite genera. Findings indicated that vertical inheritance is the primary force shaping the termite gut microbiome, with diet playing a more subtle role changing relative abundance of some populations. This suggested that gut community and structure may change in response to dietary changes as a short-term adaptive mechanism. -

(12) Patent Application Publication (10) Pub. No.: US 2013/0273277 A1 Lee Et Al
US 20130273277A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2013/0273277 A1 Lee et al. (43) Pub. Date: Oct. 17, 2013 (54) POWDERED POUCH AND METHOD OF (52) U.S. Cl. MAKING SAME CPC ...................................... B65D31/02 (2013.01) USPC .......................................... 428/35.2:427/201 (71) Applicant: MONOSOL, LLC., Merrillville, IN (US) (57) ABSTRACT (72) Inventors: David M. Lee, Crown Point, IN (US); Yashodhan S. Parulekar, Valparaiso, IN (US) Disclosed herein are water-soluble films and resulting pack ets including a water-soluble film coated by a powder, (73) Assignee: Monosol, LLC., Merrillville, IN (US) wherein the powder includes a mixture of a powdered lubri cant and an active agent. Optionally, the active agent may be (21) Appl. No.: 13/828,299 encapsulated, e.g. microencapsulated, for release of the active agent through mechanisms including, but not limited to, (22) Filed: Mar 14, 2013 mechanical rupture, melt, ablation, dissolution, diffusion, O O biodegradation, or pH-controlled release. Active ingredients Related U.S. Application Data described include enzymes, oils, flavors, colorants, odor (60) Provisional application No. 61/624,926, filed on Apr. absorbers, fragrances, pesticides, fertilizers, activators, acid 16, 2012. catalysts, metal catalysts, ion Scavengers, bleaches, bleach components, fabric softeners and combinations thereof. Publication Classification Examples of packet fills include laundry detergents, bleach and laundry additives, fabric care, dishwashing, hard Surface (51) Int. Cl. cleaning, beauty care, skin care, other personal care, and B65D 30/08 (2006.01) foodstuffs. Patent Application Publication Oct. 17, 2013 US 2013/0273277 A1 Interval Plot of Blank, Only Starch, Malodor + St Malodor +st, .. -

Generated by SRI International Pathway Tools Version 25.0, Authors S
Authors: Pallavi Subhraveti Ron Caspi Quang Ong Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_900086655Cyc: Paenibacillus sp. Marseille-P2472 Cellular Overview Connections between pathways are omitted for legibility. -

Heloisa Berti Gabriel
HELOISA BERTI GABRIEL Caracterização funcional de farnesil difosfato sintase/geranilgeranil difosfato sintase (FPPS/GGPPS) e 1,4-dihidroxi-2-naftoato preniltransferase (MenA) envolvidas respectivamente na via de isoprenóides e da vitamina K em Plasmodium falciparum Tese apresentada ao Programa de Pós-Graduação em Biologia da Relação Patógeno-Hospedeiro do Instituto de Ciências Biomédicas da Universidade de São Paulo, para obtenção do Título de Doutor em Ciências. São Paulo 2015 HELOISA BERTI GABRIEL Caracterização funcional de farnesil difosfato sintase/geranilgeranil difosfato sintase (FPPS/GGPPS) e 1,4-dihidroxi-2-naftoato preniltransferase (MenA) envolvidas respectivamente na via de isoprenóides e da vitamina K em Plasmodium falciparum Tese apresentada ao Programa de Pós-Graduação em Biologia da Relação Patógeno-Hospedeiro do Instituto de Ciências Biomédicas da Universidade de São Paulo, para obtenção do Título de Doutor em Ciências. Área de concentração: Biologia da Relação Patógeno- Hospedeiro Orientador: Prof. Dr. Alejandro Miguel Katzin Coorientador: Dr. Mauro Ferreira de Azevedo Versão Original São Paulo 2015 DADOS DE CATALOGAÇÃO NA PUBLICAÇÃO (CIP) Serviço de Biblioteca e Informação Biomédica do Instituto de Ciências Biomédicas da Universidade de São Paulo © reprodução total Gabriel, Heloisa Berti. Caracterização funcional de farnesil difosfato sintase/geranilgeranil difosfato sintase (FPPS/GGPPS) e 1,4-dihidroxi-2-naftoato preniltransferase (MenA) envolvidas respectivamente na via de isoprenóides e da vitamina K em Plasmodium