Deamination of 6‑Aminodeoxyfutalosine in Menaquinone Biosynthesis by Distantly Related Enzymes † ∥ # † ‡ § ⊥ ¶ Alissa M
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Downloaded 10/5/2021 9:43:05 AM
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Aromatic side-chain flips orchestrate the conformational sampling of functional loops in Cite this: Chem. Sci.,2021,12,9318 † All publication charges for this article human histone deacetylase 8 have been paid for by the Royal Society a a bcd of Chemistry Vaibhav Kumar Shukla, ‡ Lucas Siemons, ‡ Francesco L. Gervasio and D. Flemming Hansen *a Human histone deacetylase 8 (HDAC8) is a key hydrolase in gene regulation and an important drug-target. High-resolution structures of HDAC8 in complex with substrates or inhibitors are available, which have provided insights into the bound state of HDAC8 and its function. Here, using long all-atom unbiased molecular dynamics simulations and Markov state modelling, we show a strong correlation between the conformation of aromatic side chains near the active site and opening and closing of the surrounding functional loops of HDAC8. We also investigated two mutants known to allosterically downregulate the enzymatic activity of HDAC8. Based on experimental data, we hypothesise that I19S-HDAC8 is unable to Received 6th April 2021 Creative Commons Attribution 3.0 Unported Licence. release the product, whereas both product release and substrate binding are impaired in the S39E- Accepted 27th May 2021 HDAC8 mutant. The presented results deliver detailed insights into the functional dynamics of HDAC8 DOI: 10.1039/d1sc01929e and provide a mechanism for the substantial downregulation caused by allosteric mutations, including rsc.li/chemical-science a disease causing one. Introduction II (HDAC-4, -5, -6, -7, -9, and HDAC10), and class III (SIRT1-7) have sequence similarity to yeast Rpd3, Hda1, and Sir2, Acetylation of lysine side chains occurs as a co-translation or respectively, whereas class IV (HDAC11) shares sequence simi- 2 This article is licensed under a post-translational modication of proteins and was rst iden- larity with both class I and II proteins. -

Structural Insights Into Histone Modifying Enzymes
Wayne State University Wayne State University Theses January 2019 Structural Insights Into Histone Modifying Enzymes Shruti Amle Wayne State University, [email protected] Follow this and additional works at: https://digitalcommons.wayne.edu/oa_theses Part of the Biochemistry Commons, and the Molecular Biology Commons Recommended Citation Amle, Shruti, "Structural Insights Into Histone Modifying Enzymes" (2019). Wayne State University Theses. 693. https://digitalcommons.wayne.edu/oa_theses/693 This Open Access Embargo is brought to you for free and open access by DigitalCommons@WayneState. It has been accepted for inclusion in Wayne State University Theses by an authorized administrator of DigitalCommons@WayneState. STRUCTURAL INSIGHTS INTO HISTONE MODIFYING ENZYMES by SHRUTI AMLE THESIS Submitted to the Graduate School of Wayne State University, Detroit, Michigan in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE 2019 MAJOR: BIOCHEMISTRY AND MOLECULAR BIOLOGY Approved By: _________________________________________ Advisor Date _________________________________________ _________________________________________ _________________________________________ i ACKNOWLEDGEMENTS Writing this thesis has been extremely captivating and gratifying. I take this opportunity to express my deep and sincere acknowledgements to the number of people for extending their generous support and unstinted help during my entire study. Firstly, I would like to express my respectful regards and deep sense of gratitude to my advisor, Dr. Zhe Yang. I am extremely honored to study and work under his guidance. His vision, ideals, timely motivation and immense knowledge had a deep influence on my entire journey of this career. Without his understanding and support, it would not have been possible to complete this research successfully. I also owe my special thanks to my committee members: Dr. -

TETRAHYDROBIOPTERIN DEFICIENCY and BRAIN NITRIC OXIDE METABOLISM by Michael Peter Brand
TETRAHYDROBIOPTERIN DEFICIENCY AND BRAIN NITRIC OXIDE METABOLISM By Michael Peter Brand A thesis submitted as partial fulfilment for the degree of Doctor of Philosophy in the Faculty of Science at the University of London. March 1997 Institute of Neurology Department of Neurochemistry Queen Square London WCIN 3BG ProQuest Number: 10055824 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest. ProQuest 10055824 Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC. ProQuest LLC 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106-1346 Abstract. Tetrahydrobiopterin is an essential cofactor for the aromatic amino acid mono oxygenase group of enzymes. Inborn errors of tetrahydrobiopterin metabolism result in hyperphenylalaninaemia and impaired catecholamine and serotonin turnover. Tetrahydrobiopterin is also a cofactor for all known isoforms of nitric oxide synthase (NOS). The effect of tetrahydrobiopterin deficiency on brain nitric oxide metabolism has to date been given little consideration. In this thesis the effect of tetrahydrobiopterin deficiency on brain nitric oxide metabolism has been studied using a mouse model of tetrahydrobiopterin deficiency, the hph-1 mouse. Tetrahydrobiopterin was measured in 10 and 30 day old mice in whole brain and cerebellum. -

Structural Properties of the Nickel Ions in Urease: Novel Insights Into the Catalytic and Inhibition Mechanisms
Coordination Chemistry Reviews 190–192 (1999) 331–355 www.elsevier.com/locate/ccr Structural properties of the nickel ions in urease: novel insights into the catalytic and inhibition mechanisms Stefano Ciurli a,*, Stefano Benini b, Wojciech R. Rypniewski b, Keith S. Wilson c, Silvia Miletti a, Stefano Mangani d a Institute of Agricultural Chemistry, Uni6ersity of Bologna, Viale Berti Pichat 10, I-40127 Bologna, Italy b European Molecular Biology Laboratory, c/o DESY, Notkestraße 85, D-22603 Hamburg, Germany c Department of Chemistry, Uni6ersity of York, Heslington, York YO15DD, UK d Department of Chemistry, Uni6ersity of Siena, Pian dei Mantellini 44, I-53100 Siena, Italy Accepted 13 March 1999 Contents Abstract.................................................... 331 1. Biological background ......................................... 332 2. Spectroscopic investigations of the urease active site structure .................. 333 3. Crystallographic studies of the native enzyme ............................ 334 4. Crystallographic studies of urease mutants.............................. 341 5. Crystallographic studies of urease–inhibitor complexes ...................... 345 6. Crystallographic study of a transition state analogue bound to urease.............. 348 7. A novel proposal for the urease mechanism ............................. 350 References .................................................. 353 Abstract This work provides a comprehensive critical summary of urease spectroscopy, crystallogra- phy, inhibitor binding, and site-directed -
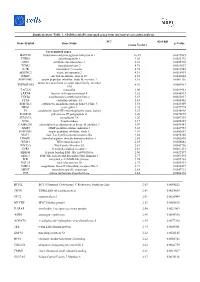
Supplementary Table 1. All Differentially Expressed Genes from Microarray Screening Analysis
Supplementary Table 1. All differentially expressed genes from microarray screening analysis. FCa (Gal-KD Gene Symbol Gene Name p-Value versus Vector) Up-regulated genes HAPLN1 hyaluronan and proteoglycan link protein 1 10.49 0.0027085 THBS1 thrombospondin 1 9.20 0.0022192 ODC1 ornithine decarboxylase 1 6.38 0.0055776 TGM2 transglutaminase 2 4.76 0.0015627 IL7R interleukin 7 receptor 4.75 0.0017245 SERINC2 serine incorporator 2 4.51 0.0014919 ITM2C integral membrane protein 2C 4.32 0.0044644 SERPINB7 serpin peptidase inhibitor, clade B, member 7 4.18 0.0081136 tumor necrosis factor receptor superfamily, member TNFRSF10D 4.01 0.0085561 10d TAGLN transgelin 3.90 0.0099963 LRRN4 leucine rich repeat neuronal 4 3.82 0.0046513 TGFB2 transforming growth factor beta 2 3.51 0.0035017 CPA4 carboxypeptidase A4 3.43 0.0008452 EPB41L3 erythrocyte membrane protein band 4.1-like 3 3.34 0.0025309 NRG1 neuregulin 1 3.28 0.0079724 F3 coagulation factor III (thromboplastin, tissue factor) 3.27 0.0038968 POLR3G polymerase III polypeptide G 3.26 0.0070675 SEMA7A semaphorin 7A 3.20 0.0087335 NT5E 5-nucleotidase 3.17 0.0036353 CAMK2N1 calmodulin-dependent protein kinase II inhibitor 1 3.07 0.0090141 TIMP3 TIMP metallopeptidase inhibitor 3 3.03 0.0047953 SERPINE1 serpin peptidase inhibitor, clade E 2.97 0.0053652 MALL mal, T-cell differentiation protein-like 2.88 0.0078205 DDAH1 dimethylarginine dimethylaminohydrolase 1 2.86 0.0002895 WDR3 WD repeat domain 3 2.85 0.0058842 WNT5A Wnt Family Member 5A 2.81 0.0043796 GPR1 G protein-coupled receptor 1 2.81 0.0021313 -

Biochemical Characterization and Comparison of Aspartylglucosaminidases Secreted in Venom of the Parasitoid Wasps Asobara Tabida and Leptopilina Heterotoma
RESEARCH ARTICLE Biochemical characterization and comparison of aspartylglucosaminidases secreted in venom of the parasitoid wasps Asobara tabida and Leptopilina heterotoma Quentin Coulette1, SeÂverine Lemauf2, Dominique Colinet2, Geneviève PreÂvost1, Caroline Anselme1, Marylène Poirie 2, Jean-Luc Gatti2* a1111111111 1 Unite ªEcologie et Dynamique des Systèmes AnthropiseÂsº (EDYSAN, FRE 3498 CNRS-UPJV), Universite de Picardie Jules Verne, Amiens, France, 2 Universite CoÃte d'Azur, INRA, CNRS, ISA, Sophia Antipolis, a1111111111 France a1111111111 a1111111111 * [email protected] a1111111111 Abstract Aspartylglucosaminidase (AGA) is a low-abundance intracellular enzyme that plays a key OPEN ACCESS role in the last stage of glycoproteins degradation, and whose deficiency leads to human Citation: Coulette Q, Lemauf S, Colinet D, PreÂvost aspartylglucosaminuria, a lysosomal storage disease. Surprisingly, high amounts of AGA- G, Anselme C, Poirie M, et al. (2017) Biochemical characterization and comparison of like proteins are secreted in the venom of two phylogenetically distant hymenopteran para- aspartylglucosaminidases secreted in venom of the sitoid wasp species, Asobara tabida (Braconidae) and Leptopilina heterotoma (Cynipidae). parasitoid wasps Asobara tabida and Leptopilina These venom AGAs have a similar domain organization as mammalian AGAs. They share heterotoma. PLoS ONE 12(7): e0181940. https:// with them key residues for autocatalysis and activity, and the mature - and -subunits also doi.org/10.1371/journal.pone.0181940 α β form an (αβ)2 structure in solution. Interestingly, only one of these AGAs subunits (α for Editor: Erjun Ling, Institute of Plant Physiology and AtAGA and for LhAGA) is glycosylated instead of the two subunits for lysosomal human Ecology Shanghai Institutes for Biological β Sciences, CHINA AGA (hAGA), and these glycosylations are partially resistant to PGNase F treatment. -

Advances in Biotechnology July 10-12, 2017 Dubai, UAE
Yu-Chen Hu, Adv Biochem Biotehcnol 2017, 02: 05 (Suppl) http://dx.doi.org/10.29011/2574-7258.C1-003 International Conference on Advances in Biotechnology July 10-12, 2017 Dubai, UAE Baculovirus-mediated MIR-214 suppression shifts osteoporotic ASCS differentiation towards osteogenesis and improves osteoporotic bone defects repair Yu-Chen Hu*, Kuei-Chang Li, Mu-Nung Hsu and Mei-Wei Lin National Tsing Hua University, Hsinchu, Taiwan Osteoporotic patients often suffer from bone fracture but its healing is compromised due to impaired osteogenesis potential of bone marrow-derived mesenchymal stem cells (BMSCs). Here we aimed to exploit adipose-derived stem cells from ovariectomized (OVX) rats (OVX-ASCs) for bone healing. We unraveled that OVX-ASCs highly expressed miR-214 and identified 2 miR-214 targets: CTNNB1 (b-catenin) and TAB2. We demonstrated that miR-214 targeting of these two genes blocked the Wnt pathway, led to preferable adipogenesis and attenuated osteogenesis, undermined the osteogenesis of co- cultured OVX-BMSCs, enhanced exosomal miR-214 release and altered cytokine secretion. As a result, OVX-ASCs implantation into OVX rats failed to heal critical-size metaphyseal bone defects. However, using hybrid baculoviruses expressing miR-214 sponges to transduce OVX-ASCs, we successfully suppressed miR-214 levels, activated the Wnt pathway, upregulated osteogenic factors -catenin/Runx2, downregulated adipogenic factors PPAR-g and C/EBP-a, shifted the differentiation propensity towards osteogenic lineage, enhanced the osteogenesis of co-cultured OVX-BMSCs, elevated BMP7/osteoprotegerin secretion and hindered exosomal miR-214/osteopontin release. Consequently, implanting the miR-214 sponge-expressing OVX-ASCs tremendously improved bone healing in OVX rats. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

Biosynthesis of Natural Products Containing Β-Amino Acids
Natural Product Reports Biosynthesis of natural products containing β -amino acids Journal: Natural Product Reports Manuscript ID: NP-REV-01-2014-000007.R1 Article Type: Review Article Date Submitted by the Author: 21-Apr-2014 Complete List of Authors: Kudo, Fumitaka; Tokyo Institute Of Technology, Department of Chemistry Miyanaga, Akimasa; Tokyo Institute Of Technology, Department of Chemistry Eguchi, T; Tokyo Institute Of Technology, Department of Chemistry and Materials Science Page 1 of 20 Natural Product Reports NPR RSC Publishing REVIEW Biosynthesis of natural products containing βββ- amino acids Cite this: DOI: 10.1039/x0xx00000x Fumitaka Kudo, a Akimasa Miyanaga, a and Tadashi Eguchi *b Received 00th January 2014, We focus here on β-amino acids as components of complex natural products because the presence of β-amino acids Accepted 00th January 2014 produces structural diversity in natural products and provides characteristic architectures beyond that of ordinary DOI: 10.1039/x0xx00000x α-L-amino acids, thus generating significant and unique biological functions in nature. In this review, we first survey the known bioactive β-amino acid-containing natural products including nonribosomal peptides, www.rsc.org/ macrolactam polyketides, and nucleoside-β-amino acid hybrids. Next, the biosynthetic enzymes that form β-amino acids from α-amino acids and de novo synthesis of β-amino acids are summarized. Then, the mechanisms of β- amino acid incorporation into natural products are reviewed. Because it is anticipated that the rational swapping of the β-amino acid moieties with various side chains and stereochemistries by biosynthetic engineering should lead to the creation of novel architectures and bioactive compounds, the accumulation of knowledge regarding β- amino acid-containing natural product biosynthetic machinery could have a significant impact in this field. -

Supplementary Information
Supplementary information (a) (b) Figure S1. Resistant (a) and sensitive (b) gene scores plotted against subsystems involved in cell regulation. The small circles represent the individual hits and the large circles represent the mean of each subsystem. Each individual score signifies the mean of 12 trials – three biological and four technical. The p-value was calculated as a two-tailed t-test and significance was determined using the Benjamini-Hochberg procedure; false discovery rate was selected to be 0.1. Plots constructed using Pathway Tools, Omics Dashboard. Figure S2. Connectivity map displaying the predicted functional associations between the silver-resistant gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S3. Connectivity map displaying the predicted functional associations between the silver-sensitive gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S4. Metabolic overview of the pathways in Escherichia coli. The pathways involved in silver-resistance are coloured according to respective normalized score. Each individual score represents the mean of 12 trials – three biological and four technical. Amino acid – upward pointing triangle, carbohydrate – square, proteins – diamond, purines – vertical ellipse, cofactor – downward pointing triangle, tRNA – tee, and other – circle. -

Active Site Tyrosine Is Essential for Amidohydrolase but Not for Esterase
Active site tyrosine is essential for amidohydrolase but not for esterase activity of a class 2 histone deacetylase-like bacterial enzyme Kristin Moreth, Daniel Riester, Christian Hildmann, René Hempel, Dennis Wegener, Andreas Schober, Andreas Schwienhorst To cite this version: Kristin Moreth, Daniel Riester, Christian Hildmann, René Hempel, Dennis Wegener, et al.. Ac- tive site tyrosine is essential for amidohydrolase but not for esterase activity of a class 2 histone deacetylase-like bacterial enzyme. Biochemical Journal, Portland Press, 2006, 401 (3), pp.659-665. 10.1042/BJ20061239. hal-00478649 HAL Id: hal-00478649 https://hal.archives-ouvertes.fr/hal-00478649 Submitted on 30 Apr 2010 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Biochemical Journal Immediate Publication. Published on 12 Oct 2006 as manuscript BJ20061239 Active site tyrosine is essential for amidohydrolase but not for esterase activity of a class 2 histone deacetylase-like bacterial enzyme Kristin Moreth¶, Daniel Riester¶, Christian Hildmann¶, René Hempel¶, Dennis Wegener¶,§,‡, -

PURINE SALVAGE in HELICOBACTER PYLORI by ERICA FRANCESCA MILLER (Under the Direction of Robert J. Maier) ABSTRACT Purines Are Es
PURINE SALVAGE IN HELICOBACTER PYLORI by ERICA FRANCESCA MILLER (Under the Direction of Robert J. Maier) ABSTRACT Purines are essential for all living cells. This fact is reflected in the high degree of pathway conservation for purine metabolism across all domains of life. The availability of purines within a mammalian host is thought to be a limiting factor for infection, as demonstrated by the importance of purine synthesis and salvage genes among many bacterial pathogens. Helicobacter pylori, a primary causative agent of peptic ulcers and gastric cancers, colonizes a niche that is otherwise uninhabited by bacteria: the surface of the human gastric epithelium. Despite many studies over the past 30 years that have addressed virulence mechanisms such as acid resistance, little knowledge exists regarding this organism’s purine metabolism. To fill this gap in knowledge, we asked whether H. pylori can carry out de novo purine biosynthesis, and whether its purine salvage network is complete. Based on genomic data from the fully sequenced H. pylori genomes, we combined mutant analysis with physiological studies to determine that H. pylori, by necessity, must acquire purines from its human host. Furthermore, we found the purine salvage network to be complete, allowing this organism to use any single purine nucleobase or nucleoside for growth. In the process of elucidating these pathways, we discovered a nucleoside transporter in H. pylori that, in contrast to the biochemically- characterized homolog NupC, aids in uptake of purine rather than pyrimidine nucleosides into the cell. Lastly, we investigated an apparent pathway gap in the genome annotation—that of adenine degradation—and in doing so uncovered a new family of adenosine deaminase that lacks sequence homology with all other adenosine deaminases studied to date.