Dissection of Heme Binding to Plasmodium Falciparum Glyceraldehyde-3-Phosphate Dehydrogenase Using Spectroscopic Methods and Molecular Docking
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Download English-US Transcript (PDF)
MITOCW | watch?v=56vQ0S2eAjw SPEAKER 1: The following content is provided under a Creative Commons license. Your support will help MIT OpenCourseWare continue to offer high quality educational resources for free. To make a donation or view additional materials from hundreds of MIT courses, visit MIT OpenCourseWare at ocw.mit.edu. PROFESSOR: Today what I want to do within the lexicon is tell you about nature's most spectacularly beautiful cofactors. And these are formed from vitamin B-12, which you find in your vitamin bottle. OK. So what is the structure of vitamin B-12, and why do I say they are spectacularly beautiful? So it's very hard to see, but if you look at the structure of this, where have you seen a molecule this complicated with five membered rings, each of which has a nitrogen in this? You've seen this when you studied hemoglobin, and you think about heme and proto protoporphyrin IX. If you look at the biosynthetic pathway of heme, a branchpoint of that pathway is to make this ring, which is found in adenosylcobalamin and methylcobalamin, which is what we're going to be focusing on today. And this ring is called the corrin ring. So what I want to do is introduce you a little bit to this corrin ring and what's unusual about it compared to protoporphyrin IX that you've seen before. So the vitamin, as in the case of all vitamins that we've talked about over the course of the semester, is not the actual cofactor used in the enzymatic transformation. -

Iron and Chelation in Biochemistry and Medicine: New Approaches to Controlling Iron Metabolism and Treating Related Diseases
cells Review Iron and Chelation in Biochemistry and Medicine: New Approaches to Controlling Iron Metabolism and Treating Related Diseases George J. Kontoghiorghes * and Christina N. Kontoghiorghe Postgraduate Research Institute of Science, Technology, Environment and Medicine, CY-3021 Limassol, Cyprus * Correspondence: [email protected]; Tel./Fax: +357-2627-2076 Received: 7 May 2020; Accepted: 5 June 2020; Published: 12 June 2020 Abstract: Iron is essential for all living organisms. Many iron-containing proteins and metabolic pathways play a key role in almost all cellular and physiological functions. The diversity of the activity and function of iron and its associated pathologies is based on bond formation with adjacent ligands and the overall structure of the iron complex in proteins or with other biomolecules. The control of the metabolic pathways of iron absorption, utilization, recycling and excretion by iron-containing proteins ensures normal biologic and physiological activity. Abnormalities in iron-containing proteins, iron metabolic pathways and also other associated processes can lead to an array of diseases. These include iron deficiency, which affects more than a quarter of the world’s population; hemoglobinopathies, which are the most common of the genetic disorders and idiopathic hemochromatosis. Iron is the most common catalyst of free radical production and oxidative stress which are implicated in tissue damage in most pathologic conditions, cancer initiation and progression, neurodegeneration and many other diseases. The interaction of iron and iron-containing proteins with dietary and xenobiotic molecules, including drugs, may affect iron metabolic and disease processes. Deferiprone, deferoxamine, deferasirox and other chelating drugs can offer therapeutic solutions for most diseases associated with iron metabolism including iron overload and deficiency, neurodegeneration and cancer, the detoxification of xenobiotic metals and most diseases associated with free radical pathology. -
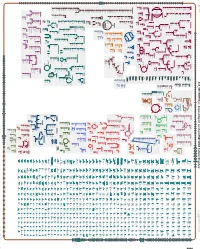
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Quang Ong Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_900114035Cyc: Amycolatopsis sacchari DSM 44468 Cellular Overview Connections between pathways are omitted for legibility. -

Definition of the Catalytic Site of Cytochrome C Oxidase: Specific Ligands of Heme a and the Heme A3-CUB Center JAMES P
Proc. Natl. Acad. Sci. USA Vol. 89, pp. 4786-4790, June 1992 Biochemistry Definition of the catalytic site of cytochrome c oxidase: Specific ligands of heme a and the heme a3-CUB center JAMES P. SHAPLEIGH*, JONATHAN P. HOSLERt, MARY M. J. TECKLENBURGO§, YOUNKYOO KIMt, GERALD T. BABCOCKt, ROBERT B. GENNIS*, AND SHELAGH FERGUSON-MILLERt *School of Chemical Sciences, University of Illinois, Urbana, IL 61801; and Departments of tBiochemistry and tChemistry, Michigan State University, East Lansing, MI 48824 Communicated by N. Edward Tolbert, February 6, 1992 (receivedfor review November 17, 1991) ABSTRACT The three-subunit aa3-type cytochrome c ox- and deduced amino acid sequences are very similar to those idase (EC 1.9.3.1) of Rhodobacter sphaeroides is structurally from the closely related bacterium Paracoccus denitrificans and functionally homologous to the more complex mitochon- (2, 10-12). In addition, subunit I shows 50% sequence drial oxidase. The largest subunit, subunit I, is highly con- identity to subunit I ofbovine heart cytochrome c oxidase (9). served and predicted to contain 12 transmembrane segments Hydropathy profile analysis of subunit I from Rb. sphae- that provide all the ligands for three of the four metal centers: roides is consistent with 12 transmembrane helical spans, as heme a, heme a3, and CUB. A variety of spectroscopic tech- previously proposed for bovine subunit I (11). The two- niques identify these ligands as histidines. We have used dimensional model that emerges from this analysis (Fig. 1) site-directed mutagenesis to change all the conserved histidines highlights seven histidine residues, six of which are found to within subunit I of cytochrome c oxidase from Rb. -

Biological Chemistry I, Lexicon
Chemistry 5.07, Fall 2013 Lexicon of biochemical reactions Topic Description Page # # 1 Review of electrophiles and nucleophiles found in Biochemistry 2 2 C-C bond formation via carbonyl chem (aldol and Claisen reactions): Aldehydes, ketones, and Coenzyme 3-5 A and thioesters 3 Prenyl transfer reactions (will not be covered in 5.07) 6 4 Redox cofactors – Flavins (FAD, FMN, riboflavin) and NAD+ (NADP+) 7-12 5 ATP and phosphoryl transfer reactions 13-15 6 Thiamine pyrophosphate 16-18 7 Lipoic acid 19 8 S-Adenosylmethionine (SAM, Adomet), methylations 20 9 Pyridoxal phosphate 21-24 10 Biotin 25 11 Hemes 26-27 12 Metal cofactors: FeS clusters, Cu clusters 28 13 Coenzyme Q 29 14 ET theory 30-32 15 Folates 33-34 16 Methylcobalamin and adenosylcobalamin 35-37 1. Review of electrophiles and nucleophiles in biology Electrophiles Nucleophiles Nucleophilic form H+ Protons + Hydroxyl group Mn+ Metal ions + Sulfhydryl group + Carbonyl carbon Amino group Imine (protonated imine) carbon + Imidazole group (Schiff base) 2 2. Carbonyl Chemistry: Mechanisms of C-C Bond Formation • There are three general ways to form C-C bonds: 1. Aldol reactions where an aldehyde is condensed in a reversible reaction with another aldehyde or a ketone; 2. Claisen reaction in which a Coenzyme A thioester is condensed with an aldehyde or a ketone; 3. Prenyl transfer reactions where two 5 carbon units are condensed to form isoprenoids. • The aldol and claisen reactions involve carbonyl chemistry. These reactions are prevalent in glycolysis, fatty acid biosynthesis and degradation and the pentose phosphate pathway. 1. Aldol reaction generalizations: a. -

Heme Deficiency May Be a Factor in the Mitochondrial and Neuronal Decay of Aging
Heme deficiency may be a factor in the mitochondrial and neuronal decay of aging Hani Atamna*, David W. Killilea, Alison Nisbet Killilea, and Bruce N. Ames* Children’s Hospital Oakland Research Institute, Oakland, CA 94609 Contributed by Bruce N. Ames, September 26, 2002 Heme, a major functional form of iron in the cell, is synthesized in other environmental toxins (15, 16). Ferrochelatase is inacti- the mitochondria by ferrochelatase inserting ferrous iron into vated when its iron-sulfur cluster disassembles because intracel- protoporphyrin IX. Heme deficiency was induced with N-methyl- lular iron levels are low (17) or nitric oxide (NO) is present (18). protoporphyrin IX, a selective inhibitor of ferrochelatase, in two Iron deficiency is by far the most studied; it impairs cognitive human brain cell lines, SHSY5Y (neuroblastoma) and U373 (astro- function in children, impacts the morphology and the physiology cytoma), as well as in rat primary hippocampal neurons. Heme of brain even before changes in hemoglobin appear (19), dam- deficiency in brain cells decreases mitochondrial complex IV, acti- ages mitochondria (20), and causes oxidative stress (20). Iron vates nitric oxide synthase, alters amyloid precursor protein, and deficiency in children is associated with difficulties in performing corrupts iron and zinc homeostasis. The metabolic consequences cognitive tasks and retardation in the development of the CNS resulting from heme deficiency seem similar to dysfunctional (21, 22). Prenatal iron deficiency causes loss of cytochrome c neurons in patients with Alzheimer’s disease. Heme-deficient oxidase (complex IV) in selected regions in the brain of rats (23) SHSY5Y or U373 cells die when induced to differentiate or to and causes a decrease in ferrochelatase (17). -

Structural Basis of Inter-Domain Electron Transfer in Ncb5or, a Redox Enzyme Implicated in Diabetes and Lipid Metabolism
STRUCTURAL BASIS OF INTER-DOMAIN ELECTRON TRANSFER IN NCB5OR, A REDOX ENZYME IMPLICATED IN DIABETES AND LIPID METABOLISM By Bin Deng Submitted to the graduate degree program in Rehabilitation Science and the Graduate Faculty of the University of Kansas in partial fulfillment of the requirements for the degree of Doctor of Philosophy Hao Zhu, Ph.D. (co-chair, advisor) Irina Smirnova, Ph.D. (co-chair) David Benson, Ph.D. (co-advisor) WenFang Wang, Ph.D. Aron Fenton, Ph.D. Date defended: August 17, 2011 The Dissertation Committee for Bin Deng certifies that this is the approved version of the following dissertation STRUCTURAL BASIS OF INTER-DOMAIN ELECTRON TRANSFER IN NCB5OR, A REDOX ENZYME IMPLICATED IN DIABETES AND LIPID METABOLISM Hao Zhu, Ph.D. (co-chair, advisor) Irina Smirnova, Ph.D. (co-chair) Date approved: August 22, 2011 ii ABSTRACT NADH cytochrome b5 oxidoreductase (Ncb5or) is a multi-domain redox enzyme found in all animal tissues and associated with the endoplasmic reticulum (ER). Ncb5or contains (from N-terminus to C terminus) a novel N-terminal region, the b5 domain (Ncb5or-b5), the CS domain, and the b5R domain (Ncb5or-b5R). Ncb5or-b5, the heme binding domain, is homologous to microsomal cytochrome b5 (Cyb5A) and belongs to cytochrome b5 superfamily. Ncb5or-b5R, the FAD (flavin adenine dinucleotide) binding domain, is homologous to cytochrome b5 reductase (Cyb5R3) and belongs to ferredoxin NADP+ reductase superfamily. Both superfamilies are of great biological significance whose members have important functions. The CS domain can be assigned into the heat shock protein 20 (HSP20, or p23) family, whose members are known to mediate protein-protein interactions. -

Significance of Heme and Heme Degradation in the Pathogenesis Of
International Journal of Molecular Sciences Review Significance of Heme and Heme Degradation in the Pathogenesis of Acute Lung and Inflammatory Disorders Stefan W. Ryter Proterris, Inc., Boston, MA 02118, USA; [email protected] Abstract: The heme molecule serves as an essential prosthetic group for oxygen transport and storage proteins, as well for cellular metabolic enzyme activities, including those involved in mitochondrial respiration, xenobiotic metabolism, and antioxidant responses. Dysfunction in both heme synthesis and degradation pathways can promote human disease. Heme is a pro-oxidant via iron catalysis that can induce cytotoxicity and injury to the vascular endothelium. Additionally, heme can modulate inflammatory and immune system functions. Thus, the synthesis, utilization and turnover of heme are by necessity tightly regulated. The microsomal heme oxygenase (HO) system degrades heme to carbon monoxide (CO), iron, and biliverdin-IXα, that latter which is converted to bilirubin-IXα by biliverdin reductase. Heme degradation by heme oxygenase-1 (HO-1) is linked to cytoprotection via heme removal, as well as by activity-dependent end-product generation (i.e., bile pigments and CO), and other potential mechanisms. Therapeutic strategies targeting the heme/HO-1 pathway, including therapeutic modulation of heme levels, elevation (or inhibition) of HO-1 protein and activity, and application of CO donor compounds or gas show potential in inflammatory conditions including sepsis and pulmonary diseases. Keywords: acute lung injury; carbon monoxide; heme; heme oxygenase; inflammation; lung dis- ease; sepsis Citation: Ryter, S.W. Significance of Heme and Heme Degradation in the Pathogenesis of Acute Lung and Inflammatory Disorders. Int. J. Mol. 1. Introduction Sci. -

Designer Fungus FAD Glucose Dehydrogenase Capable of Direct Electron Transfer T
Biosensors and Bioelectronics 123 (2019) 114–123 Contents lists available at ScienceDirect Biosensors and Bioelectronics journal homepage: www.elsevier.com/locate/bios Designer fungus FAD glucose dehydrogenase capable of direct electron transfer T Kohei Itoa, Junko Okuda-Shimazakib, Kazushige Morib, Katsuhiro Kojimab, Wakako Tsugawaa, ⁎ Kazunori Ikebukuroa, Chi-En Linc,Jeffrey La Bellec, Hiromi Yoshidad, Koji Sodea,b,e, a Department of Biotechnology and Life Science, Graduate School of Engineering, Tokyo University of Agriculture and Technology, 2-24-16 Naka-cho, Koganei, Tokyo 184- 8588, Japan b Ultizyme International Ltd., 1-13-16, Minami, Meguro, Tokyo 152-0013, Japan c School of Biological and Health System Engineering, Ira A. Fulton Schools of Engineering, Arizona State University, P.O. Box 879709, Tempe, AZ 85287-9719, USA d Life Science Research Center and Faculty of Medicine, Kagawa University, 1750-1 Ikenobe, Miki-cho, Kita-gun, Kagawa 761-0793, Japan e Joint Department of Biomedical Engineering, The University of North Carolina at Chapel Hill and North Carolina State University, Chapel Hill, NC 27599, USA ARTICLE INFO ABSTRACT Keywords: Fungi-derived flavin adenine dinucleotide glucose dehydrogenases (FADGDHs) are currently the most popular Glucose dehydrogenase and advanced enzymes for self-monitoring of blood glucose sensors; however, the achievement of direct electron Cellobiose dehydrogenase transfer (DET) with FADGDHs is difficult. In this study, a designer FADGDH was constructed by fusing Aspergillus Direct electron transfer flavus derived FADGDH (AfGDH) and a Phanerochaete chrisosporium CDH (PcCDH)-derived heme b-binding cy- Designer FADGDH tochrome domain to develop a novel FADGDH that is capable of direct electron transfer with an electrode. -

GAPDH Regulates Cellular Heme Insertion Into Inducible Nitric Oxide Synthase
GAPDH regulates cellular heme insertion into inducible nitric oxide synthase Ritu Chakravartia, Kulwant S. Aulaka, Paul L. Foxb, and Dennis J. Stuehra,1 aDepartment of Pathobiology, and bDepartment of Cell Biology, Lerner Research institute, Cleveland Clinic Foundation, Cleveland, OH 44195 Edited by Solomon H. Snyder, The Johns Hopkins University School of Medicine, Baltimore, MD, and approved September 10, 2010 (received for review June 11, 2010) Heme proteins play essential roles in biology, but little is known NOS enzymes, hemoglobin, catalase, and cytochrome P450 (17). about heme transport inside mammalian cells or how heme is Importantly, the mechanism of NO action did not involve inserted into soluble proteins. We recently found that nitric oxide changes in heme availability or effects of NO on cellular energy (NO) blocks cells from inserting heme into several proteins, production or activation of soluble guanylate cyclase (17). The including cytochrome P450s, hemoglobin, NO synthases, and results implied that NO might act on an uncharacterized yet catalase. This finding led us to explore the basis for NO inhibition common cellular component or process to generally inhibit heme and to identify cytosolic proteins that may be involved, using insertion into proteins. This concept served as a starting point to inducible NO synthase (iNOS) as a model target. Surprisingly, we search for proteins that might drive and regulate intracellular found that GAPDH plays a key role. GAPDH was associated with heme insertion reactions in mammals. In our current study, we iNOS in cells. Pure GAPDH bound tightly to heme or to iNOS in an used iNOS as a model heme protein target, and identify a cyto- solic protein that binds heme, functions in heme insertion, and is NO-sensitive manner. -

Hc-Hrg-2, a Glutathione Transferase Gene, Regulates Heme Homeostasis
Zhou et al. Parasites Vectors (2020) 13:40 https://doi.org/10.1186/s13071-020-3911-z Parasites & Vectors RESEARCH Open Access Hc‑hrg‑2, a glutathione transferase gene, regulates heme homeostasis in the blood-feeding parasitic nematode Haemonchus contortus Jing‑Ru Zhou1, Dan‑Ru Bu1, Xian‑Feng Zhao2, Fei Wu1, Xue‑Qiu Chen1, Heng‑Zhi Shi1, Chao‑Qun Yao3, Ai‑Fang Du1* and Yi Yang1* Abstract Background: Haemonchus contortus, a blood‑feeding parasite, is constantly surrounded by large quantities of heme released from the catabolism of host red blood cells. To cope with the toxicity of free heme, H. contortus needs to uptake and detoxify the heme, a process believed to be paramount for parasite survival. Methods: A heme‑responsive gene Hc-hrg-2 was identifed which is the homologue of Ce-hrg-2. The transcriptional levels in all developmental stages and heme‑responsive ability of Hc-hrg-2 were analyzed by qRT‑PCR. Immunofuo‑ rescence analysis and cell transfections were performed to analyze the expression pattern of Hc‑HGR‑2. Statistical analyses were performed with GraghPad Prism 6.0 using Student’s t‑test. Results: To investigate the heme homeostasis of H. contortus, we frst identifed a heme‑responsive gene Hc-hrg-2, a homolog of Ce-hrg-2 that is involved in heme transport in the hypodermis of Caenorhabditis elegans. Using qRT‑PCR, we showed that Hc-hrg-2 mRNA was expressed throughout all life‑cycle stages of H. contortus with the highest level in the third‑stage larvae (L3s). Notably, transcription of Hc-hrg-2 in the exsheathed L3s was signifcantly upregulated in the presence of high concentration of heme. -

Polyamines Mitigate Antibiotic Inhibition of A.Actinomycetemcomitans Growth
Polyamines Mitigate Antibiotic Inhibition of A.actinomycetemcomitans Growth THESIS Presented in Partial Fulfillment of the Requirements for the Degree Master of Science in the Graduate School of The Ohio State University By Allan Wattimena Graduate Program in Dentistry The Ohio State University 2017 Master's Examination Committee: Dr John Walters, Advisor Dr Purnima Kumar Dr Sara Palmer Dr Shareef Dabdoub Copyright by Allan Wattimena 2017 Abstract Polyamines are ubiquitous polycationic molecules that are present in all prokaryotic and eukaryotic cells. They are the breakdown products of amino acids and are important modulators of cell growth, stress and cell proliferation. Polyamines are present in higher concentrations in the periodontal pocket and may affect antibiotic resistance of bacterial biofilms. The effect of polyamines was investigated with amoxicillin (AMX), azithromycin (AZM) and doxycycline (DOX) on the growth of Aggregatibacter actinomycetemcomitans (A.a.) Y4 strain. Bacteria were grown in brain heart infusion broth under the following conditions: 1) A.a. only, 2) A.a. + antibiotic, 3) A.a. + antibiotic + polyamine mix (1.4mM putrescine, 0.4mM spermidine, 0.4mM spermine). Growth curve analysis, MIC determination and metatranscriptomic analysis were carried out. The presence of exogenous polyamines produced a small, but significant increase in growth of A.a. Polyamines mitigated the inhibitory effect of AMX, AZM and DOX on A.a. growth. Metatranscriptomic analysis revealed differing transcriptomic profiles when comparing AMX and AZM in the presence of polyamines. Polyamines produced a transient mitigation of AMX inhibition, but did not have a significant effect on gene transcription. Many gene transcription changes were seen when polyamines were in the presence of AZM.