Destruction of Biological Tetrapyrrole Macrocycles by Hypochlorous Acid and Its Scavenging by Lycopene Dhiman Maitra Wayne State University
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Magnesium-Protoporphyrin Chelatase of Rhodobacter
Proc. Natl. Acad. Sci. USA Vol. 92, pp. 1941-1944, March 1995 Biochemistry Magnesium-protoporphyrin chelatase of Rhodobacter sphaeroides: Reconstitution of activity by combining the products of the bchH, -I, and -D genes expressed in Escherichia coli (protoporphyrin IX/tetrapyrrole/chlorophyll/bacteriochlorophyll/photosynthesis) LUCIEN C. D. GIBSON*, ROBERT D. WILLOWSt, C. GAMINI KANNANGARAt, DITER VON WETTSTEINt, AND C. NEIL HUNTER* *Krebs Institute for Biomolecular Research and Robert Hill Institute for Photosynthesis, Department of Molecular Biology and Biotechnology, University of Sheffield, Sheffield, S10 2TN, United Kingdom; and tCarlsberg Laboratory, Department of Physiology, Gamle Carlsberg Vej 10, DK-2500 Copenhagen Valby, Denmark Contributed by Diter von Wettstein, November 14, 1994 ABSTRACT Magnesium-protoporphyrin chelatase lies at Escherichia coli and demonstrate that the extracts of the E. coli the branch point of the heme and (bacterio)chlorophyll bio- transformants can convert Mg-protoporphyrin IX to Mg- synthetic pathways. In this work, the photosynthetic bacte- protoporphyrin monomethyl ester (20, 21). Apart from posi- rium Rhodobacter sphaeroides has been used as a model system tively identifying bchM as the gene encoding the Mg- for the study of this reaction. The bchH and the bchI and -D protoporphyrin methyltransferase, this work opens up the genes from R. sphaeroides were expressed in Escherichia coli. possibility of extending this approach to other parts of the When cell-free extracts from strains expressing BchH, BchI, pathway. In this paper, we report the expression of the genes and BchD were combined, the mixture was able to catalyze the bchH, -I, and -D from R. sphaeroides in E. coli: extracts from insertion of Mg into protoporphyrin IX in an ATP-dependent these transformants, when combined in vitro, are highly active manner. -

Download English-US Transcript (PDF)
MITOCW | watch?v=56vQ0S2eAjw SPEAKER 1: The following content is provided under a Creative Commons license. Your support will help MIT OpenCourseWare continue to offer high quality educational resources for free. To make a donation or view additional materials from hundreds of MIT courses, visit MIT OpenCourseWare at ocw.mit.edu. PROFESSOR: Today what I want to do within the lexicon is tell you about nature's most spectacularly beautiful cofactors. And these are formed from vitamin B-12, which you find in your vitamin bottle. OK. So what is the structure of vitamin B-12, and why do I say they are spectacularly beautiful? So it's very hard to see, but if you look at the structure of this, where have you seen a molecule this complicated with five membered rings, each of which has a nitrogen in this? You've seen this when you studied hemoglobin, and you think about heme and proto protoporphyrin IX. If you look at the biosynthetic pathway of heme, a branchpoint of that pathway is to make this ring, which is found in adenosylcobalamin and methylcobalamin, which is what we're going to be focusing on today. And this ring is called the corrin ring. So what I want to do is introduce you a little bit to this corrin ring and what's unusual about it compared to protoporphyrin IX that you've seen before. So the vitamin, as in the case of all vitamins that we've talked about over the course of the semester, is not the actual cofactor used in the enzymatic transformation. -

AOP 131: Aryl Hydrocarbon Receptor Activation Leading to Uroporphyria
Organisation for Economic Co-operation and Development DOCUMENT CODE For Official Use English - Or. English 1 January 1990 AOP 131: Aryl hydrocarbon receptor activation leading to uroporphyria Short Title: AHR activation-uroporphyria This document was approved by the Extended Advisory Group on Molecular Screening and Toxicogenomics in June 2018. The Working Group of the National Coordinators of the Test Guidelines Programme and the Working Party on Hazard Assessment are invited to review and endorse the AOP by 29 March 2019. Magdalini Sachana, Administrator, Hazard Assessment, [email protected], +(33- 1) 85 55 64 23 Nathalie Delrue, Administrator, Test Guidelines, [email protected], +(33-1) 45 24 98 44 This document, as well as any data and map included herein, are without prejudice to the status of or sovereignty over any territory, to the delimitation of international frontiers and boundaries and to the name of any territory, city or area. 2 │ Foreword This Adverse Outcome Pathway (AOP) on Aryl hydrocarbon receptor activation leading to uroporphyria, has been developed under the auspices of the OECD AOP Development Programme, overseen by the Extended Advisory Group on Molecular Screening and Toxicogenomics (EAGMST), which is an advisory group under the Working Group of the National Coordinators for the Test Guidelines Programme (WNT). The AOP has been reviewed internally by the EAGMST, externally by experts nominated by the WNT, and has been endorsed by the WNT and the Working Party on Hazard Assessment (WPHA) in xxxxx. Through endorsement of this AOP, the WNT and the WPHA express confidence in the scientific review process that the AOP has undergone and accept the recommendation of the EAGMST that the AOP be disseminated publicly. -

Iron and Chelation in Biochemistry and Medicine: New Approaches to Controlling Iron Metabolism and Treating Related Diseases
cells Review Iron and Chelation in Biochemistry and Medicine: New Approaches to Controlling Iron Metabolism and Treating Related Diseases George J. Kontoghiorghes * and Christina N. Kontoghiorghe Postgraduate Research Institute of Science, Technology, Environment and Medicine, CY-3021 Limassol, Cyprus * Correspondence: [email protected]; Tel./Fax: +357-2627-2076 Received: 7 May 2020; Accepted: 5 June 2020; Published: 12 June 2020 Abstract: Iron is essential for all living organisms. Many iron-containing proteins and metabolic pathways play a key role in almost all cellular and physiological functions. The diversity of the activity and function of iron and its associated pathologies is based on bond formation with adjacent ligands and the overall structure of the iron complex in proteins or with other biomolecules. The control of the metabolic pathways of iron absorption, utilization, recycling and excretion by iron-containing proteins ensures normal biologic and physiological activity. Abnormalities in iron-containing proteins, iron metabolic pathways and also other associated processes can lead to an array of diseases. These include iron deficiency, which affects more than a quarter of the world’s population; hemoglobinopathies, which are the most common of the genetic disorders and idiopathic hemochromatosis. Iron is the most common catalyst of free radical production and oxidative stress which are implicated in tissue damage in most pathologic conditions, cancer initiation and progression, neurodegeneration and many other diseases. The interaction of iron and iron-containing proteins with dietary and xenobiotic molecules, including drugs, may affect iron metabolic and disease processes. Deferiprone, deferoxamine, deferasirox and other chelating drugs can offer therapeutic solutions for most diseases associated with iron metabolism including iron overload and deficiency, neurodegeneration and cancer, the detoxification of xenobiotic metals and most diseases associated with free radical pathology. -
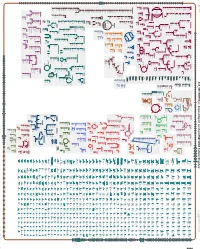
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Quang Ong Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_900114035Cyc: Amycolatopsis sacchari DSM 44468 Cellular Overview Connections between pathways are omitted for legibility. -

On Tuning the Fluorescence Emission of Porphyrin Free Bases Bonded to the Pore Walls of Organo-Modified Silica
Molecules 2014, 19, 2261-2285; doi:10.3390/molecules19022261 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Article On Tuning the Fluorescence Emission of Porphyrin Free Bases Bonded to the Pore Walls of Organo-Modified Silica Rosa I. Y. Quiroz-Segoviano 1, Iris N. Serratos 1, Fernando Rojas-González 1, Salvador R. Tello-Solís 1, Rebeca Sosa-Fonseca 2, Obdulia Medina-Juárez 1, Carmina Menchaca-Campos 3 and Miguel A. García-Sánchez 1,* 1 Departamento de Química, Universidad Autónoma Metropolitana-Iztapalapa, Av. San Rafael Atlixco 186, Vicentina, D. F. 09340, Mexico 2 Departamento de Física, Universidad Autónoma Metropolitana-Iztapalapa, Av. San Rafael Atlixco 186, Vicentina, D. F. 09340, Mexico 3 Centro de Investigación en Ingeniería y Ciencias Aplicadas, UAEM, Av. Universidad 1001, Col. Chamilpa, C.P. 62209, Cuernavaca Mor., Mexico * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +52-55-5804-4677; Fax: +52-55-5804-4666. Received: 24 December 2013; in revised form: 29 January 2014 / Accepted: 7 February 2014 / Published: 21 February 2014 Abstract: A sol-gel methodology has been duly developed in order to perform a controlled covalent coupling of tetrapyrrole macrocycles (e.g., porphyrins, phthalocyanines, naphthalocyanines, chlorophyll, etc.) to the pores of metal oxide networks. The resulting absorption and emission spectra intensities in the UV-VIS-NIR range have been found to depend on the polarity existing inside the pores of the network; in turn, this polarization can be tuned through the attachment of organic substituents to the tetrapyrrrole macrocycles before bonding them to the pore network. -

Definition of the Catalytic Site of Cytochrome C Oxidase: Specific Ligands of Heme a and the Heme A3-CUB Center JAMES P
Proc. Natl. Acad. Sci. USA Vol. 89, pp. 4786-4790, June 1992 Biochemistry Definition of the catalytic site of cytochrome c oxidase: Specific ligands of heme a and the heme a3-CUB center JAMES P. SHAPLEIGH*, JONATHAN P. HOSLERt, MARY M. J. TECKLENBURGO§, YOUNKYOO KIMt, GERALD T. BABCOCKt, ROBERT B. GENNIS*, AND SHELAGH FERGUSON-MILLERt *School of Chemical Sciences, University of Illinois, Urbana, IL 61801; and Departments of tBiochemistry and tChemistry, Michigan State University, East Lansing, MI 48824 Communicated by N. Edward Tolbert, February 6, 1992 (receivedfor review November 17, 1991) ABSTRACT The three-subunit aa3-type cytochrome c ox- and deduced amino acid sequences are very similar to those idase (EC 1.9.3.1) of Rhodobacter sphaeroides is structurally from the closely related bacterium Paracoccus denitrificans and functionally homologous to the more complex mitochon- (2, 10-12). In addition, subunit I shows 50% sequence drial oxidase. The largest subunit, subunit I, is highly con- identity to subunit I ofbovine heart cytochrome c oxidase (9). served and predicted to contain 12 transmembrane segments Hydropathy profile analysis of subunit I from Rb. sphae- that provide all the ligands for three of the four metal centers: roides is consistent with 12 transmembrane helical spans, as heme a, heme a3, and CUB. A variety of spectroscopic tech- previously proposed for bovine subunit I (11). The two- niques identify these ligands as histidines. We have used dimensional model that emerges from this analysis (Fig. 1) site-directed mutagenesis to change all the conserved histidines highlights seven histidine residues, six of which are found to within subunit I of cytochrome c oxidase from Rb. -

Biological Chemistry I, Lexicon
Chemistry 5.07, Fall 2013 Lexicon of biochemical reactions Topic Description Page # # 1 Review of electrophiles and nucleophiles found in Biochemistry 2 2 C-C bond formation via carbonyl chem (aldol and Claisen reactions): Aldehydes, ketones, and Coenzyme 3-5 A and thioesters 3 Prenyl transfer reactions (will not be covered in 5.07) 6 4 Redox cofactors – Flavins (FAD, FMN, riboflavin) and NAD+ (NADP+) 7-12 5 ATP and phosphoryl transfer reactions 13-15 6 Thiamine pyrophosphate 16-18 7 Lipoic acid 19 8 S-Adenosylmethionine (SAM, Adomet), methylations 20 9 Pyridoxal phosphate 21-24 10 Biotin 25 11 Hemes 26-27 12 Metal cofactors: FeS clusters, Cu clusters 28 13 Coenzyme Q 29 14 ET theory 30-32 15 Folates 33-34 16 Methylcobalamin and adenosylcobalamin 35-37 1. Review of electrophiles and nucleophiles in biology Electrophiles Nucleophiles Nucleophilic form H+ Protons + Hydroxyl group Mn+ Metal ions + Sulfhydryl group + Carbonyl carbon Amino group Imine (protonated imine) carbon + Imidazole group (Schiff base) 2 2. Carbonyl Chemistry: Mechanisms of C-C Bond Formation • There are three general ways to form C-C bonds: 1. Aldol reactions where an aldehyde is condensed in a reversible reaction with another aldehyde or a ketone; 2. Claisen reaction in which a Coenzyme A thioester is condensed with an aldehyde or a ketone; 3. Prenyl transfer reactions where two 5 carbon units are condensed to form isoprenoids. • The aldol and claisen reactions involve carbonyl chemistry. These reactions are prevalent in glycolysis, fatty acid biosynthesis and degradation and the pentose phosphate pathway. 1. Aldol reaction generalizations: a. -

Heme Deficiency May Be a Factor in the Mitochondrial and Neuronal Decay of Aging
Heme deficiency may be a factor in the mitochondrial and neuronal decay of aging Hani Atamna*, David W. Killilea, Alison Nisbet Killilea, and Bruce N. Ames* Children’s Hospital Oakland Research Institute, Oakland, CA 94609 Contributed by Bruce N. Ames, September 26, 2002 Heme, a major functional form of iron in the cell, is synthesized in other environmental toxins (15, 16). Ferrochelatase is inacti- the mitochondria by ferrochelatase inserting ferrous iron into vated when its iron-sulfur cluster disassembles because intracel- protoporphyrin IX. Heme deficiency was induced with N-methyl- lular iron levels are low (17) or nitric oxide (NO) is present (18). protoporphyrin IX, a selective inhibitor of ferrochelatase, in two Iron deficiency is by far the most studied; it impairs cognitive human brain cell lines, SHSY5Y (neuroblastoma) and U373 (astro- function in children, impacts the morphology and the physiology cytoma), as well as in rat primary hippocampal neurons. Heme of brain even before changes in hemoglobin appear (19), dam- deficiency in brain cells decreases mitochondrial complex IV, acti- ages mitochondria (20), and causes oxidative stress (20). Iron vates nitric oxide synthase, alters amyloid precursor protein, and deficiency in children is associated with difficulties in performing corrupts iron and zinc homeostasis. The metabolic consequences cognitive tasks and retardation in the development of the CNS resulting from heme deficiency seem similar to dysfunctional (21, 22). Prenatal iron deficiency causes loss of cytochrome c neurons in patients with Alzheimer’s disease. Heme-deficient oxidase (complex IV) in selected regions in the brain of rats (23) SHSY5Y or U373 cells die when induced to differentiate or to and causes a decrease in ferrochelatase (17). -

Preparation of Tetrapyrrole-Amino Acid Covalent Complexes
I'lunt I'ht.siol.Ritx ltt'nt. 1996. -14 (3). 393-39lt Preparation of tetrapyrrole-amino acid covalent complexes Leszek Fiedorl'2*, Varda Rosenbach-Belkinl, Maruthi Sail and Avigdor Scherzl I BiochernistryDepartment. The Weizn-rannInstitute of Science.76100 Rehovot.Israel. I Prcscntaddress: Institute of Molecular BiologSr.Ja-ciellonian University. Al. Mickiewicza 3. 3 l- 120 Cracow. Poland. ':'Author to whom correspondenceshould bc addrcsscd(fax +48-12-336907:E-mail fiedor@)mol.uj.edu.pl) Abstract The presentedsynthetic approach towards chcn'rical modifications of chlorophylls(Chls) provides a perspectivcto construct model systems. where tetrapyrrole-aminoacid and tetrapyrrole-peptideinteractions coulcl be studied in covalent rnodel compor,rncls. The approach relies on thc lact that in Chls the | 7r propionic rcid sidc chain docs not participatc in the tetrapl'rroleii--electron system. It makes use of a plant enzvmechlolophyllase (EC 3.1.1.1,+).which lrr lilo and in yitrc catalysesreactions at this sidc function. The transesterilicationand hyclrolysisenzymatic rerctions are useful on a preparativescale. ln the transesterificationreaction. a desiredamino acid rcsiduc posscssirrgprimary hydloxyl group can be directly attachedto the propiorric acid side chain o1' Chl. This mcthod allows to replace the phytyl moiety in Chls n'ith seline. The r:rtherreaction. enzyrratic hydrolysis of Chls, yields chlorophyllides and opens a convenientroutc fbr furthcr rnodifications.If sufliciently mild synthetic mcthodsarc uscd. such as catalysisw,ith ,l-dimethyl arnino pyridine or activationwith N-hvdroxvsuccinimide.an arrino acid or peptide residuecan be covalentlybound to chlorophyllides' carboxylic group. lear,'ingthe essentialclectlonic structure of Chl intact. The activation w'ith N-hydroxvsuccininridcallows fbr the coupling cvrn in aqueous rncdia. -

Structural Basis of Inter-Domain Electron Transfer in Ncb5or, a Redox Enzyme Implicated in Diabetes and Lipid Metabolism
STRUCTURAL BASIS OF INTER-DOMAIN ELECTRON TRANSFER IN NCB5OR, A REDOX ENZYME IMPLICATED IN DIABETES AND LIPID METABOLISM By Bin Deng Submitted to the graduate degree program in Rehabilitation Science and the Graduate Faculty of the University of Kansas in partial fulfillment of the requirements for the degree of Doctor of Philosophy Hao Zhu, Ph.D. (co-chair, advisor) Irina Smirnova, Ph.D. (co-chair) David Benson, Ph.D. (co-advisor) WenFang Wang, Ph.D. Aron Fenton, Ph.D. Date defended: August 17, 2011 The Dissertation Committee for Bin Deng certifies that this is the approved version of the following dissertation STRUCTURAL BASIS OF INTER-DOMAIN ELECTRON TRANSFER IN NCB5OR, A REDOX ENZYME IMPLICATED IN DIABETES AND LIPID METABOLISM Hao Zhu, Ph.D. (co-chair, advisor) Irina Smirnova, Ph.D. (co-chair) Date approved: August 22, 2011 ii ABSTRACT NADH cytochrome b5 oxidoreductase (Ncb5or) is a multi-domain redox enzyme found in all animal tissues and associated with the endoplasmic reticulum (ER). Ncb5or contains (from N-terminus to C terminus) a novel N-terminal region, the b5 domain (Ncb5or-b5), the CS domain, and the b5R domain (Ncb5or-b5R). Ncb5or-b5, the heme binding domain, is homologous to microsomal cytochrome b5 (Cyb5A) and belongs to cytochrome b5 superfamily. Ncb5or-b5R, the FAD (flavin adenine dinucleotide) binding domain, is homologous to cytochrome b5 reductase (Cyb5R3) and belongs to ferredoxin NADP+ reductase superfamily. Both superfamilies are of great biological significance whose members have important functions. The CS domain can be assigned into the heat shock protein 20 (HSP20, or p23) family, whose members are known to mediate protein-protein interactions. -

Continuous Chlorophyll Degradation Accompanied by Chlorophyllide and Phytol Reutilization for Chlorophyll Synthesis in Synechocystis Sp
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Biochimica et Biophysica Acta 1767 (2007) 920–929 www.elsevier.com/locate/bbabio Continuous chlorophyll degradation accompanied by chlorophyllide and phytol reutilization for chlorophyll synthesis in Synechocystis sp. PCC 6803 ⁎ Dmitrii Vavilin, Wim Vermaas School of Life Sciences and Center for the Study of Early Events in Photosynthesis, Arizona State University, Box 874501, Tempe, AZ 85287, USA Received 3 January 2007; received in revised form 23 March 2007; accepted 27 March 2007 Available online 3 April 2007 Abstract Chlorophyll synthesis and degradation were analyzed in the cyanobacterium Synechocystis sp. PCC 6803 by incubating cells in the presence of 13C-labeled glucose or 15N-containing salts. Upon mass spectral analysis of chlorophyll isolated from cells grown in the presence of 13C-glucose for different time periods, four chlorophyll pools were detected that differed markedly in the amount of 13C incorporated into the porphyrin (Por) and phytol (Phy) moieties of the molecule. These four pools represent (i) unlabeled chlorophyll (12Por12Phy), (ii) 13C-labeled chlorophyll (13Por13Phy), and (iii, iv) chlorophyll, in which either the porphyrin or the phytol moiety was 13C-labeled, whereas the other constituent of the molecule remained unlabeled (13Por12Phy and 12Por13Phy). The kinetics of 12Por12Phy disappearance, presumably due to chlorophyll de- esterification, and of 13Por12Phy, 12Por13Phy, and 13Por13Phy accumulation due to chlorophyll synthesis provided evidence for continuous chlorophyll turnover in Synechocystis cells. The loss of 12Por12Phy was three-fold faster in a photosystem I-less strain than in a photosystem II- less strain and was accelerated in wild-type cells upon exposure to strong light.