Operando Infrared Spectroscopy for the Analysis of Gas-Processing Metalloenzymes Sven T
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Reduction by the Methylreductase System in Methanobacterium Bryantii WILLIAM B
JOURNAL OF BACTERIOLOGY, Jan. 1987, p. 87-92 Vol. 169, No. 1 0021-9193/87/010087-06$02.00/0 Copyright © 1987, American Society for Microbiology Inhibition by Corrins of the ATP-Dependent Activation and CO2 Reduction by the Methylreductase System in Methanobacterium bryantii WILLIAM B. WHITMAN'* AND RALPH S. WOLFE2 Department of Microbiology, University of Georgia, Athens, Georgia 30602,1 and Department of Microbiology, University ofIllinois, Urbana, Illinois 618012 Received 1 August 1986/Accepted 28 September 1986 Corrins inhibited the ATP-dependent activation of the methylreductase system and the methyl coenzyme M-dependent reduction of CO2 in extracts of Methanobacterium bryantii resolved from low-molecular-weight factors. The concentrations of cobinamides and cobamides required for one-half of maximal inhibition of the ATP-depen4ent activation were between 1 and 5 ,M. Cobinamides were more inhibitory at lower concentra- tiops than cobamides. Deoxyadenosylcobalamin was not inhibitory at concentrations up to 25 ,uM. The inhibition of CO2 reduction was competitive with respect to CO2. The concentration of methylcobalamin required for one-half of maximal inhibition was 5 ,M. Other cobamideg inhibited at similar concentrations, but diaquacobinami4e inhibited at lower concentrations. With respect to their affinities and specificities for corrins, inhibition of both the ATP-dependent activation'and CO2 reduction closely resembled the corrin- dependent activation of the methylreductase described in similar extracts (W. B. Whitman and R. S. Wolfe, J. Bacteriol. 164:165-172, 1985). However, whether the multiple effects of corrins are due to action at a single site is unknown. The effect of corrins (cobamides and cobinamides) on in CO2 reduction. -

The Wolfe Cycle Comes Full Circle
The Wolfe cycle comes full circle Rudolf K. Thauer1 Max Planck Institute for Terrestrial Microbiology, D-35043 Marburg, Germany n 1988, Rouvière and Wolfe (1) H - ΔμNa+ 2 CO2 suggested that methane formation + MFR from H and CO by methanogenic + 2H+ *Fd + H O I 2 2 ox 2 archaea could be a cyclical process. j O = Indirect evidence indicated that the CoB-SH + CoM-SH fi *Fd 2- a rst step, the reduction of CO2 to for- red R mylmethanofuran, was somehow coupled + * H MPT 2 H2 Fdox 4 to the last step, the reduction of the het- h erodisulfide (CoM-S-S-CoB) to coenzyme CoM-S-S-CoB b MFR M (CoM-SH) and coenzyme B (CoB-SH). H Over 2 decades passed until the coupling C 4 10 mechanism was unraveled in 2011: Via g flavin-based electron bifurcation, the re- CoB-SH duction of CoM-S-S-CoB with H provides 2 H+ the reduced ferredoxin (Fig. 1h) required c + Purines for CO2 reduction to formylmethanofuran ΔμNa + H MPT 4 f H O (2) (Fig. 1a). However, one question still 2 remained unanswered: How are the in- termediates replenished that are removed CoM-SH for the biosynthesis of cell components H Methionine d from CO2 (orange arrows in Fig. 1)? This Acetyl-CoA e anaplerotic (replenishing) reaction has F420 F420H2 recently been identified by Lie et al. (3) as F420 F420H2 the sodium motive force-driven reduction H i of ferredoxin with H2 catalyzed by the i energy-converting hydrogenase EhaA-T H2 (green arrow in Fig. -

Annotation Guidelines for Experimental Procedures
Annotation Guidelines for Experimental Procedures Developed By Mohammed Alliheedi Robert Mercer Version 1 April 14th, 2018 1- Introduction and background information What is rhetorical move? A rhetorical move can be defined as a text fragment that conveys a distinct communicative goal, in other words, a sentence that implies an author’s specific purpose to readers. What are the types of rhetorical moves? There are several types of rhetorical moves. However, we are interested in 4 rhetorical moves that are common in the method section of a scientific article that follows the Introduction Methods Results and Discussion (IMRaD) structure. 1- Description of a method: It is concerned with a sentence(s) that describes experimental events (e.g., “Beads with bound proteins were washed six times (for 10 min under rotation at 4°C) with pulldown buffer and proteins harvested in SDS-sample buffer, separated by SDS-PAGE, and analyzed by autoradiography.” (Ester & Uetz, 2008)). 2- Appeal to authority: It is concerned with a sentence(s) that discusses the use of standard methods, protocols, and procedures. There are two types of this move: - A reference to a well-established “standard” method (e.g., the use of a method like “PCR” or “electrophoresis”). - A reference to a method that was previously described in the literature (e.g., “Protein was determined using fluorescamine assay [41].” (Larsen, Frandesn and Treiman, 2001)). 3- Source of materials: It is concerned with a sentence(s) that lists the source of biological materials that are used in the experiment (e.g., “All microalgal strains used in this study are available at the Elizabeth Aidar Microalgae Culture Collection, Department of Marine Biology, Federal Fluminense University, Brazil.” (Larsen, Frandesn and Treiman, 2001)). -

Untargetted Metabolomic Exploration of the Mycobacterium Tuberculosis Stress Response to Cinnamon Essential Oil
biomolecules Article Untargetted Metabolomic Exploration of the Mycobacterium tuberculosis Stress Response to Cinnamon Essential Oil Elwira Sieniawska 1,* , Rafał Sawicki 2 , Joanna Golus 2 and Milen I. Georgiev 3,4 1 Chair and Department of Pharmacognosy, Medical University of Lublin, Chodzki 1, 20-093 Lublin, Poland 2 Chair and Department of Biochemistry and Biotechnology, Medical University of Lublin, Chodzki 1, 20-093 Lublin, Poland; [email protected] (R.S.); [email protected] (J.G.) 3 Group of Plant Cell Biotechnology and Metabolomics, The Stephan Angeloff Institute of Microbiology, Bulgarian Academy of Sciences, 139 Ruski Blvd., 4000 Plovdiv, Bulgaria; [email protected] 4 Center of Plant Systems Biology and Biotechnology, 4000 Plovdiv, Bulgaria * Correspondence: [email protected] Received: 6 January 2020; Accepted: 24 February 2020; Published: 26 February 2020 Abstract: The antimycobacterial activity of cinnamaldehyde has already been proven for laboratory strains and for clinical isolates. What is more, cinnamaldehyde was shown to threaten the mycobacterial plasma membrane integrity and to activate the stress response system. Following promising applications of metabolomics in drug discovery and development we aimed to explore the mycobacteria response to cinnamaldehyde within cinnamon essential oil treatment by untargeted liquid chromatography–mass spectrometry. The use of predictive metabolite pathway analysis and description of produced lipids enabled the evaluation of the stress symptoms shown by bacteria. This study suggests that bacteria exposed to cinnamaldehyde could reorganize their outer membrane as a physical barrier against stress factors. They probably lowered cell wall permeability and inner membrane fluidity, and possibly redirected carbon flow to store energy in triacylglycerols. Being a reactive compound, cinnamaldehyde may also contribute to disturbances in bacteria redox homeostasis and detoxification mechanisms. -

Towards Kinetic Modeling of Global Metabolic Networks with Incomplete Experimental Input on Kinetic Parameters
Towards Kinetic Modeling of Global Metabolic Networks with Incomplete Experimental Input on Kinetic Parameters Ping Ao 1,2 , Lik Wee Lee 1, Mary E. Lidstrom 3,4 , Lan Yin 5, and Xiaomei Zhu 6 1 Department of Mechanical Engineering , University of Washington , Seattle , WA 98195, USA 2 Department of Physics , University of Washington , Seattle , WA 98195, USA 3 Department of Microbiology , University of Washington , Seattle , WA 98195, USA 4 Department of Chemical Engineering , University of Washington , Seattle , WA 98195, USA 5 School of Physics , Peking University , 100871 Beijing , PR China 6 GeneMath , 5525 27 th Ave . N .E., Seattle , WA 98105, USA Abstract: This is the first report on a systematic method for constructing a large scale kinetic metabolic model and its initial application to the modeling of central metabolism of Methylobacterium extorquens AM1, a methylotrophic and environmental important bacterium. Its central metabolic network includes formaldehyde metabolism, serine cycle, citric acid cycle, pentose phosphate pathway, gluconeogensis, PHB synthesis and acetyl-CoA conversion pathway, respiration and energy metabolism. Through a systematic and consistent procedure of finding a set of parameters in the physiological range we overcome an outstanding difficulty in large scale kinetic modeling: the requirement for a massive number of enzymatic reaction parameters. We are able to construct the kinetic model based on general biological considerations and incomplete experimental kinetic parameters. Our method consists of -

Recycling of Vitamin B12 and NAD+ Within the Pdu Microcompartment of Salmonella Enterica Shouqiang Cheng Iowa State University
Iowa State University Capstones, Theses and Graduate Theses and Dissertations Dissertations 2010 Recycling of vitamin B12 and NAD+ within the Pdu microcompartment of Salmonella enterica Shouqiang Cheng Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/etd Part of the Biochemistry, Biophysics, and Structural Biology Commons Recommended Citation Cheng, Shouqiang, "Recycling of vitamin B12 and NAD+ within the Pdu microcompartment of Salmonella enterica" (2010). Graduate Theses and Dissertations. 11713. https://lib.dr.iastate.edu/etd/11713 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. + Recycling of vitamin B12 and NAD within the Pdu microcompartment of Salmonella enterica by Shouqiang Cheng A dissertation submitted to the graduate faculty in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Major: Biochemistry Program of Study Committee: Thomas A. Bobik, Major Professor Alan DiSpirito Basil Nikolau Reuben Peters Gregory J. Phillips Iowa State University Ames, Iowa 2010 Copyright © Shouqiang Cheng, 2010. All rights reserved. ii Table of contents Abstract............................................................................................................................. -

Download English-US Transcript (PDF)
MITOCW | watch?v=56vQ0S2eAjw SPEAKER 1: The following content is provided under a Creative Commons license. Your support will help MIT OpenCourseWare continue to offer high quality educational resources for free. To make a donation or view additional materials from hundreds of MIT courses, visit MIT OpenCourseWare at ocw.mit.edu. PROFESSOR: Today what I want to do within the lexicon is tell you about nature's most spectacularly beautiful cofactors. And these are formed from vitamin B-12, which you find in your vitamin bottle. OK. So what is the structure of vitamin B-12, and why do I say they are spectacularly beautiful? So it's very hard to see, but if you look at the structure of this, where have you seen a molecule this complicated with five membered rings, each of which has a nitrogen in this? You've seen this when you studied hemoglobin, and you think about heme and proto protoporphyrin IX. If you look at the biosynthetic pathway of heme, a branchpoint of that pathway is to make this ring, which is found in adenosylcobalamin and methylcobalamin, which is what we're going to be focusing on today. And this ring is called the corrin ring. So what I want to do is introduce you a little bit to this corrin ring and what's unusual about it compared to protoporphyrin IX that you've seen before. So the vitamin, as in the case of all vitamins that we've talked about over the course of the semester, is not the actual cofactor used in the enzymatic transformation. -

Methane Utilization in Methylomicrobium Alcaliphilum 20ZR: a Systems Approach Received: 8 September 2017 Ilya R
www.nature.com/scientificreports OPEN Methane utilization in Methylomicrobium alcaliphilum 20ZR: a systems approach Received: 8 September 2017 Ilya R. Akberdin1,2, Merlin Thompson1, Richard Hamilton1, Nalini Desai3, Danny Alexander3, Accepted: 22 January 2018 Calvin A. Henard4, Michael T. Guarnieri4 & Marina G. Kalyuzhnaya1 Published: xx xx xxxx Biological methane utilization, one of the main sinks of the greenhouse gas in nature, represents an attractive platform for production of fuels and value-added chemicals. Despite the progress made in our understanding of the individual parts of methane utilization, our knowledge of how the whole-cell metabolic network is organized and coordinated is limited. Attractive growth and methane-conversion rates, a complete and expert-annotated genome sequence, as well as large enzymatic, 13C-labeling, and transcriptomic datasets make Methylomicrobium alcaliphilum 20ZR an exceptional model system for investigating methane utilization networks. Here we present a comprehensive metabolic framework of methane and methanol utilization in M. alcaliphilum 20ZR. A set of novel metabolic reactions governing carbon distribution across central pathways in methanotrophic bacteria was predicted by in-silico simulations and confrmed by global non-targeted metabolomics and enzymatic evidences. Our data highlight the importance of substitution of ATP-linked steps with PPi-dependent reactions and support the presence of a carbon shunt from acetyl-CoA to the pentose-phosphate pathway and highly branched TCA cycle. The diverged TCA reactions promote balance between anabolic reactions and redox demands. The computational framework of C1-metabolism in methanotrophic bacteria can represent an efcient tool for metabolic engineering or ecosystem modeling. Climate change concerns linked to increasing concentrations of anthropogenic methane have spiked inter- est in biological methane utilization as a novel platform for improving ecological and human well-being1–4. -

Community and Proteomic Analysis of Anaerobic Consortia Converting Tetramethylammonium to Methane
Supporting Information for Manuscript Submitted to Archaea Community and Proteomic Analysis of Anaerobic Consortia Converting Tetramethylammonium to Methane Wei-Yu Chen1, Lucia Kraková2, Jer-Horng Wu1*, Domenico Pangallo2, Lenka Jeszeová2, Bing Liu3, Hidenari Yasui3 , 1Department of Environmental Engineering, National Cheng Kung University, No.1, University Road, East District, Tainan City 701, Taiwan (ROC) 2Institute of Molecular Biology, Slovak Academy of Sciences, Dubravska cesta 21, 84551 Bratislava, Slovakia 3Faculty of Environmental Engineering, The University of Kitakyushu, 1-1, Hibikino, Wakamatsu, Kitakyushu, Japan 808-0135 1 Table S1 Annotation of proteins related to the conversion of methylamines by the methanogens in the CMJP sample analyzed in this study. Relative Coverage Peptides Accession Organism gene Annotation abundance (%) (#) (%) A0A0E3SIM3 Methanosarcina barkeri 3 MSBR3_0618 Dimethylamine:corrinoid methyltransferase 17.13 21 1.39 A0A0E3Q5F5 Methanosarcina vacuolata Z-761 MSVAZ_1366 Methanol methyltransferase corrinoid protein 12.94 15 0.28 A0A0E3P0I6 Methanosarcina siciliae T4/M MSSIT_0113 Monomethylamine:corrinoid methyltransferase 23.58 20 0.48 A0A0E3WTL8 Methanosarcina lacustris Z-7289 MSLAZ_3204 Monomethylamine:corrinoid methyltransferase 15.94 9 0.24 A0A0E3KRC1 Methanosarcina thermophila CHTI-55 MSTHC_1357 Monomethylamine:corrinoid methyltransferase 18.12 10 0.22 A0A0E3WT78 Methanosarcina lacustris Z-7289 MSLAZ_2479 Dimethylamine methyltransferase corrinoid protein 25.00 11 0.13 A0A0E3Q3C8 Methanosarcina vacuolata -

Iron and Chelation in Biochemistry and Medicine: New Approaches to Controlling Iron Metabolism and Treating Related Diseases
cells Review Iron and Chelation in Biochemistry and Medicine: New Approaches to Controlling Iron Metabolism and Treating Related Diseases George J. Kontoghiorghes * and Christina N. Kontoghiorghe Postgraduate Research Institute of Science, Technology, Environment and Medicine, CY-3021 Limassol, Cyprus * Correspondence: [email protected]; Tel./Fax: +357-2627-2076 Received: 7 May 2020; Accepted: 5 June 2020; Published: 12 June 2020 Abstract: Iron is essential for all living organisms. Many iron-containing proteins and metabolic pathways play a key role in almost all cellular and physiological functions. The diversity of the activity and function of iron and its associated pathologies is based on bond formation with adjacent ligands and the overall structure of the iron complex in proteins or with other biomolecules. The control of the metabolic pathways of iron absorption, utilization, recycling and excretion by iron-containing proteins ensures normal biologic and physiological activity. Abnormalities in iron-containing proteins, iron metabolic pathways and also other associated processes can lead to an array of diseases. These include iron deficiency, which affects more than a quarter of the world’s population; hemoglobinopathies, which are the most common of the genetic disorders and idiopathic hemochromatosis. Iron is the most common catalyst of free radical production and oxidative stress which are implicated in tissue damage in most pathologic conditions, cancer initiation and progression, neurodegeneration and many other diseases. The interaction of iron and iron-containing proteins with dietary and xenobiotic molecules, including drugs, may affect iron metabolic and disease processes. Deferiprone, deferoxamine, deferasirox and other chelating drugs can offer therapeutic solutions for most diseases associated with iron metabolism including iron overload and deficiency, neurodegeneration and cancer, the detoxification of xenobiotic metals and most diseases associated with free radical pathology. -
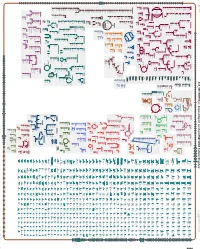
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Quang Ong Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_900114035Cyc: Amycolatopsis sacchari DSM 44468 Cellular Overview Connections between pathways are omitted for legibility. -

Definition of the Catalytic Site of Cytochrome C Oxidase: Specific Ligands of Heme a and the Heme A3-CUB Center JAMES P
Proc. Natl. Acad. Sci. USA Vol. 89, pp. 4786-4790, June 1992 Biochemistry Definition of the catalytic site of cytochrome c oxidase: Specific ligands of heme a and the heme a3-CUB center JAMES P. SHAPLEIGH*, JONATHAN P. HOSLERt, MARY M. J. TECKLENBURGO§, YOUNKYOO KIMt, GERALD T. BABCOCKt, ROBERT B. GENNIS*, AND SHELAGH FERGUSON-MILLERt *School of Chemical Sciences, University of Illinois, Urbana, IL 61801; and Departments of tBiochemistry and tChemistry, Michigan State University, East Lansing, MI 48824 Communicated by N. Edward Tolbert, February 6, 1992 (receivedfor review November 17, 1991) ABSTRACT The three-subunit aa3-type cytochrome c ox- and deduced amino acid sequences are very similar to those idase (EC 1.9.3.1) of Rhodobacter sphaeroides is structurally from the closely related bacterium Paracoccus denitrificans and functionally homologous to the more complex mitochon- (2, 10-12). In addition, subunit I shows 50% sequence drial oxidase. The largest subunit, subunit I, is highly con- identity to subunit I ofbovine heart cytochrome c oxidase (9). served and predicted to contain 12 transmembrane segments Hydropathy profile analysis of subunit I from Rb. sphae- that provide all the ligands for three of the four metal centers: roides is consistent with 12 transmembrane helical spans, as heme a, heme a3, and CUB. A variety of spectroscopic tech- previously proposed for bovine subunit I (11). The two- niques identify these ligands as histidines. We have used dimensional model that emerges from this analysis (Fig. 1) site-directed mutagenesis to change all the conserved histidines highlights seven histidine residues, six of which are found to within subunit I of cytochrome c oxidase from Rb.