1 Altered Integrin Expression Patterns Revealed by Microarray in Human
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

140503 IPF Signatures Supplement Withfigs Thorax
Supplementary material for Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis Daryle J. DePianto1*, Sanjay Chandriani1⌘*, Alexander R. Abbas1, Guiquan Jia1, Elsa N. N’Diaye1, Patrick Caplazi1, Steven E. Kauder1, Sabyasachi Biswas1, Satyajit K. Karnik1#, Connie Ha1, Zora Modrusan1, Michael A. Matthay2, Jasleen Kukreja3, Harold R. Collard2, Jackson G. Egen1, Paul J. Wolters2§, and Joseph R. Arron1§ 1Genentech Research and Early Development, South San Francisco, CA 2Department of Medicine, University of California, San Francisco, CA 3Department of Surgery, University of California, San Francisco, CA ⌘Current address: Novartis Institutes for Biomedical Research, Emeryville, CA. #Current address: Gilead Sciences, Foster City, CA. *DJD and SC contributed equally to this manuscript §PJW and JRA co-directed this project Address correspondence to Paul J. Wolters, MD University of California, San Francisco Department of Medicine Box 0111 San Francisco, CA 94143-0111 [email protected] or Joseph R. Arron, MD, PhD Genentech, Inc. MS 231C 1 DNA Way South San Francisco, CA 94080 [email protected] 1 METHODS Human lung tissue samples Tissues were obtained at UCSF from clinical samples from IPF patients at the time of biopsy or lung transplantation. All patients were seen at UCSF and the diagnosis of IPF was established through multidisciplinary review of clinical, radiological, and pathological data according to criteria established by the consensus classification of the American Thoracic Society (ATS) and European Respiratory Society (ERS), Japanese Respiratory Society (JRS), and the Latin American Thoracic Association (ALAT) (ref. 5 in main text). Non-diseased normal lung tissues were procured from lungs not used by the Northern California Transplant Donor Network. -

Proteomic Expression Profile in Human Temporomandibular Joint
diagnostics Article Proteomic Expression Profile in Human Temporomandibular Joint Dysfunction Andrea Duarte Doetzer 1,*, Roberto Hirochi Herai 1 , Marília Afonso Rabelo Buzalaf 2 and Paula Cristina Trevilatto 1 1 Graduate Program in Health Sciences, School of Medicine, Pontifícia Universidade Católica do Paraná (PUCPR), Curitiba 80215-901, Brazil; [email protected] (R.H.H.); [email protected] (P.C.T.) 2 Department of Biological Sciences, Bauru School of Dentistry, University of São Paulo, Bauru 17012-901, Brazil; [email protected] * Correspondence: [email protected]; Tel.: +55-41-991-864-747 Abstract: Temporomandibular joint dysfunction (TMD) is a multifactorial condition that impairs human’s health and quality of life. Its etiology is still a challenge due to its complex development and the great number of different conditions it comprises. One of the most common forms of TMD is anterior disc displacement without reduction (DDWoR) and other TMDs with distinct origins are condylar hyperplasia (CH) and mandibular dislocation (MD). Thus, the aim of this study is to identify the protein expression profile of synovial fluid and the temporomandibular joint disc of patients diagnosed with DDWoR, CH and MD. Synovial fluid and a fraction of the temporomandibular joint disc were collected from nine patients diagnosed with DDWoR (n = 3), CH (n = 4) and MD (n = 2). Samples were subjected to label-free nLC-MS/MS for proteomic data extraction, and then bioinformatics analysis were conducted for protein identification and functional annotation. The three Citation: Doetzer, A.D.; Herai, R.H.; TMD conditions showed different protein expression profiles, and novel proteins were identified Buzalaf, M.A.R.; Trevilatto, P.C. -

Supporting Information for Proteomics DOI 10.1002/Pmic.200401336
Supporting Information for Proteomics DOI 10.1002/pmic.200401336 Ilan Beer, Eilon Barnea, Arie Admon Centralized data analysis of a large interlaboratory proteomics project-A feasibility study ª 2005 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.de Number Number Amino Number of high IPI of acid Protein name of labs quality peptides coverage spectra IPI00022229.1 236 8 3490 59 Apolipoprotein B-100 precursor IPI00164623.2 151 8 12671 82 IPI00032257.1 150 4 12628 83 complement component 3 precursor IPI00022434.1 110 8 13599 94 Serum albumin precursor Complement C4 precursor [Contains: C4A IPI00032258.2 99 8 5449 58 anaphylatox IPI00022463.1 94 8 9905 82 Serotransferrin precursor IPI00032256.1 92 8 15676 67 Alpha-2-macroglobulin precursor IPI00328461.1 84 8 2103 51 Hypothetical protein DKFZp686H112 Complement C5 precursor [Contains: C5a IPI00032291.1 74 6 591 50 anaphylatox IPI00179357.1 68 8 20 2 Titin IPI00017601.1 66 8 3910 65 Ceruloplasmin precursor IPI00029739.1 65 8 2265 57 Splice isoform 1 of P08603 IPI00182635.1 65 8 1358 46 IPI00220327.1 56 7 4729 61 keratin 1 IPI00021841.1 51 8 7728 91 Apolipoprotein A-I precursor IPI00019580.1 51 6 701 67 Plasminogen precursor IPI00025426.1 48 4 3184 38 Pregnancy zone protein precursor IPI00021304.1 48 4 2241 68 Keratin, type II cytoskeletal 2 epidermal IPI00013933.1 48 4 59 20 Splice isoform DPI of P15924 IPI00294193.1 46 8 1916 56 Splice isoform 1 of Q14624 IPI00021885.1 45 7 1811 45 Splice isoform Alpha-E of P02671 IPI00305457.1 44 8 5917 76 Alpha-1-antitrypsin precursor -

Genome Wide Assessment of Early Osseointegration in Implant-Adherent Cells
Genome Wide assessment of Early Osseointegration in Implant-Adherent Cells Ghadeer N. Thalji A dissertation submitted to the faculty of the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the School of Dentistry (Oral Biology) Chapel Hill 2012 Approved by Lyndon F.Cooper, DDS, PhD Eric T. Everett, PhD Gustavo Mendonça, DDS, MS, PhD Salvador Nares DDS, PhD Clark M. Stanford, DDS, PhD © 2012 Ghadeer N. Thalji ALL RIGHTS RESERVED ii ABSTRACT GHADEER N. THALJI: Genome wide assessment of early osseointegration in implant-adherent cells (Under the direction of Professor Lyndon F. Cooper) Objectives: To determine the molecular processes involved in osseointegration. Materials and methods: A structured literature review concerning in vitro and in vivo molecular assessment of osseointegration was performed. A rat and a human model were then used to identify the early molecular processes involved in osseointegration associated with a micro roughened and nanosurface superimposed featured implants. In the rat model, 32 titanium implants with surface topographies exhibiting a micro roughened (AT-II) and nanosurface superimposed featured implants (AT-I) were placed in the tibiae of 8 rats and subsequently harvested at 2 and 4 days after placement. Whereas in the human model, four titanium mini-implants with either a moderately roughened surface (TiOblast) or super-imposed nanoscale topography (Osseospeed) were placed in edentulous sites of eleven systemically healthy subjects and subsequently removed after 3 and 7 days. Total RNA was isolated from cells adherent to retrieved implants. A whole genome microarray using the Affymetrix 1.1 ST Array platform was used to describe the gene expression profiles that were differentially regulated by the implant surfaces. -

Análisis De Los Perfiles De
“ANÁLISIS DE LOS PERFILES DE EXPRESIÓN DE GENES RELACIONADOS CON VÍAS DE PROLIFERACIÓN CELULAR EN FIBROBLASTOS DE CALVARIA DE UNA MUESTRA DE PACIENTES COLOMBIANOS CON SÍNDROME DE APERT POSTERIOR A LA DEGRADACIÓN HEPARÁN SULFATO” Diana Alexandra Ramírez Montaño Universidad Nacional de Colombia Facultad de Medicina Maestría en Genética Humana Bogotá, Colombia 2016 “ANÁLISIS DE LOS PERFILES DE EXPRESIÓN DE GENES RELACIONADOS CON VÍAS DE PROLIFERACIÓN CELULAR EN FIBROBLASTOS DE CALVARIA DE UNA MUESTRA DE PACIENTES COLOMBIANOS CON SÍNDROME DE APERT POSTERIOR A LA DEGRADACIÓN HEPARÁN SULFATO” Diana Alexandra Ramírez Montaño Tesis presentada como requisito parcial para optar al título de: Magister en Genética Humana Director: Harvy Mauricio Velasco Parra, MD Especialista en Genética Médica MsC Ciencias Biológicas Línea de Investigación: Anomalías congénitas Grupo de investigación: Genética Clínica Universidad Nacional de Colombia Facultad de Medicina Maestría en Genética Humana Bogotá, Colombia 2016 VII A mi madre, por ella y para ella. X Agradecimientos Agradezco inicialmente a Dios por cada paso que me ha permitido dar en mi vida.Gracias al Dr. Harvy Velasco, por convertirse desde hace varios años en mi mentor y guía en el camino de la investigación, por la confianza en mis capacidades académicas y por la paciencia durante todos estos años. A la Dra. Lilian Torres y al equipo de la Fundación Universitaria en Ciencias de la Salud por su apoyo para la realización de este proyecto. Así mismo, al Dr. Ángel Carracedo y su equipo de la Fundación Pública de Medicina Xenómica en la Universidad de Santiago de Compostela por sus aportes en el análisis de datos que permitieron la interpretación y aprovechamiento de los datos obtenidos. -

UCLA Electronic Theses and Dissertations
UCLA UCLA Electronic Theses and Dissertations Title Proteomic Analysis of Cancer Cell Metabolism Permalink https://escholarship.org/uc/item/8t36w919 Author Chai, Yang Publication Date 2013 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA Los Angeles Proteomic Analysis of Cancer Cell Metabolism A thesis submitted in partial satisfaction of the requirements of the degree Master of Science in Oral Biology by Yang Chai 2013 ABSTRACT OF THESIS Proteomic Analysis of Cancer Cell Metabolism by Yang Chai Master of Science in Oral Biology University of California, Los Angeles, 2013 Professor Shen Hu, Chair Tumor cells can adopt alternative metabolic pathways during oncogenesis. This is an event characterized by an enhanced utilization of glucose for rapid synthesis of macromolecules such as nucleotides, lipids and proteins. This phenomenon was also known as the ‘Warburg effect’, distinguished by a shift from oxidative phosphorylation to increased aerobic glycolysis in many types of cancer cells. Increased aerobic glycolysis was also indicated with enhanced lactate production and glutamine consumption, and has been suggested to confer growth advantage for proliferating cells during oncogenic transformation. Development of a tracer-based ii methodology to determine de novo protein synthesis by tracing metabolic pathways from nutrient utilization may certainly enhance current understanding of nutrient gene interaction in cancer cells. We hypothesized that the metabolic phenotype of cancer cells as characterized by nutrient utilization for protein synthesis is significantly altered during oncogenesis, and 13C stable isotope tracers may incorporate 13C into non-essential amino acids of protein peptides during de novo protein synthesis to reflect the underlying mechanisms in cancer cell metabolism. -

Cell-Deposited Matrix Improves Retinal Pigment Epithelium Survival on Aged Submacular Human Bruch’S Membrane
Retinal Cell Biology Cell-Deposited Matrix Improves Retinal Pigment Epithelium Survival on Aged Submacular Human Bruch’s Membrane Ilene K. Sugino,1 Vamsi K. Gullapalli,1 Qian Sun,1 Jianqiu Wang,1 Celia F. Nunes,1 Noounanong Cheewatrakoolpong,1 Adam C. Johnson,1 Benjamin C. Degner,1 Jianyuan Hua,1 Tong Liu,2 Wei Chen,2 Hong Li,2 and Marco A. Zarbin1 PURPOSE. To determine whether resurfacing submacular human most, as cell survival is the worst on submacular Bruch’s Bruch’s membrane with a cell-deposited extracellular matrix membrane in these eyes. (Invest Ophthalmol Vis Sci. 2011;52: (ECM) improves retinal pigment epithelial (RPE) survival. 1345–1358) DOI:10.1167/iovs.10-6112 METHODS. Bovine corneal endothelial (BCE) cells were seeded onto the inner collagenous layer of submacular Bruch’s mem- brane explants of human donor eyes to allow ECM deposition. here is no fully effective therapy for the late complications of age-related macular degeneration (AMD), the leading Control explants from fellow eyes were cultured in medium T cause of blindness in the United States. The prevalence of only. The deposited ECM was exposed by removing BCE. Fetal AMD-associated choroidal new vessels (CNVs) and/or geo- RPE cells were then cultured on these explants for 1, 14, or 21 graphic atrophy (GA) in the U.S. population 40 years and older days. The explants were analyzed quantitatively by light micros- is estimated to be 1.47%, with 1.75 million citizens having copy and scanning electron microscopy. Surviving RPE cells from advanced AMD, approximately 100,000 of whom are African explants cultured for 21 days were harvested to compare bestro- American.1 The prevalence of AMD increases dramatically with phin and RPE65 mRNA expression. -

KRT6C Gene Keratin 6C
KRT6C gene keratin 6C Normal Function The KRT6C gene provides instructions for making a protein called keratin 6c or K6c. Keratins are a group of tough, fibrous proteins that form the structural framework of certain cells, particularly cells that make up the skin, hair, and nails. Keratin 6c is found in the skin, although it is unknown which other tissues may produce this protein. Keratin 6c is a component of molecules called keratin intermediate filaments. These filaments assemble into dense networks that provide strength and resilience to the skin, nails, and other tissues. Networks of keratin intermediate filaments protect these tissues from being damaged by friction and other everyday physical stresses. Health Conditions Related to Genetic Changes Pachyonychia congenita At least four mutations in the KRT6C gene have been found to cause pachyonychia congenita, a rare condition that primarily affects the nails and skin. In most cases, this condition becomes apparent within the first few months of life. One of the mutations associated with pachyonychia congenita changes a single protein building block (amino acid) in the keratin 6c protein. Specifically, this mutation replaces the amino acid glutamic acid with the amino acid lysine at protein position 472 (written as Glu472Lys or E472K). The other KRT6C gene mutations delete one or more amino acids from the keratin 6c protein. All of the known KRT6C gene mutations alter the structure of keratin 6c and interfere with the assembly of the keratin intermediate filament network. Without this network, skin cells become fragile and are easily damaged, making the skin less resistant to friction and minor trauma. -

Identification of Potential Salivary Biomarker Panels for Oral
www.nature.com/scientificreports OPEN Identifcation of potential salivary biomarker panels for oral squamous cell carcinoma Anu Jain1, Chinmaya Narayana Kotimoole2, Sushmita Ghoshal3, Jaimanti Bakshi4, Aditi Chatterjee5,6, Thottethodi Subrahmanya Keshava Prasad2 & Arnab Pal1* Oral squamous cell carcinoma (OSCC) is one of the most prevalent cancers worldwide with the maximum number of incidences and deaths reported from India. One of the major causes of poor survival rate associated with OSCC has been attributed to late presentation due to non-availability of a biomarker. Identifcation of early diagnostic biomarker will help in reducing the disease morbidity and mortality. We validated 12 salivary proteins using targeted proteomics, identifed initially by relative quantifcation of salivary proteins on LC–MS, in OSCC patients and controls. Salivary AHSG (p = 0.0041**) and KRT6C (p = 0.002**) were upregulated in OSCC cases and AZGP1 (p ≤ 0.0001***), KLK1 (p = 0.006**) and BPIFB2 (p = 0.0061**) were downregulated. Regression modelling resulted in a signifcant risk prediction model (p < 0.0001***) consisting of AZGP1, AHSG and KRT6C for which ROC curve had AUC, sensitivity and specifcity of 82.4%, 78% and 73.5% respectively for all OSCC cases and 87.9%, 87.5% and 73.5% respectively for late stage (T3/T4) OSCC. AZGP1, AHSG, KRT6C and BPIFB2 together resulted in ROC curve (p < 0.0001***) with AUC, sensitivity and specifcity of 94%, 100% and 77.6% respectively for N0 cases while KRT6C and AZGP1 for N+ cases with ROC curve (p < 0.0001***) having AUC sensitivity and specifcity of 76.8%, 73% and 69.4%. Our data aids in the identifcation of biomarker panels for the diagnosis of OSCC cases with a diferential diagnosis between early and late- stage cases. -
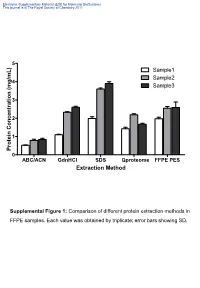
Supplemental Table 1
Electronic Supplementary Material (ESI) for Molecular BioSystems This journal is © The Royal Society of Chemistry 2011 5 Sample1 Sample2 4 Sample3 3 2 1 Protein Concentration (mg/mL) Concentration Protein 0 ABC/ACN GdnHCl SDS Qproteome FFPE PES Extraction Method Supplemental Figure 1: Comparison of different protein extraction methods in FFPE samples. Each value was obtained by triplicate; error bars showing SD. Electronic Supplementary Material (ESI) for Molecular BioSystems This journal is © The Royal Society of Chemistry 2011 1400 1200 1000 800 600 400 Protein extracted (micrograms) extracted Protein 200 0 ABC/ACN GdnHCl SDS Qproteome FFPE PES Supplemental Figure 2: Total protein extracted using different protein extraction methods in FFPE samples. Three samples were extracted. Each measurement was obtained by triplicate; error bars showing SD. Electronic Supplementary Material (ESI) for Molecular BioSystems This journal is © The Royal Society of Chemistry 2011 Molecular Function binding protein binding FF FFPE nucleotide binding structural molecule activity RNA binding cytoskeletal protein binding identical protein binding nucleoside-triphosphatase activity protein dimerization activity transcription cofactor activity protein heterodimeri zation activity structural constituent of cytoskeleton unfolded protein binding -0.8% -0.6% -0.4% -0.2% -0.0% 0% 20% 40% 60% 80% Percentage of identified proteins Percentage of identified phosphoproteins Biological Process cellular process macromolecule metabolic process FF biopolymer metabolic process -

Ovarian Cancer–Peritoneal Cell Interactions Promote Extracellular
2311 C Ricciardelli et al. Ovarian cancer–peritoneal 23:11 T155–T168 Thematic Review cell interaction WOMEN IN CANCER THEMATIC REVIEW Ovarian cancer–peritoneal cell interactions promote extracellular matrix processing 1 1 2 1,3 C Ricciardelli , N A Lokman , M P Ween and M K Oehler Correspondence 1Discipline of Obstetrics and Gynaecology, Adelaide Medical School, Robinson Research Institute, should be addressed University of Adelaide, Adelaide, South Australia, Australia to C Ricciardelli 2Lung Research Laboratory, Hanson Institute, Department of Thoracic Medicine, Royal Adelaide Hospital, Email Adelaide, South Australia, Australia carmela.ricciardelli@ 3Department of Gynaecological Oncology, Royal Adelaide Hospital, Adelaide, South Australia, Australia adelaide.edu.au Abstract Ovarian cancer has a distinct tendency for metastasising via shedding of cancerous Key Words cells into the peritoneal cavity and implanting onto the peritoneum that lines the f ovarian cancer pelvic organs. Once ovarian cancer cells adhere to the peritoneal cells, they migrate f extracellular matrix through the peritoneal layer and invade the local organs. Alterations in the extracellular f plasmin environment are critical for tumour initiation, progression and intra-peritoneal f annexin A2 dissemination. To increase our understanding of the molecular mechanisms involved in f tumour ovarian cancer metastasis and to identify novel therapeutic targets, we recently studied microenvironment Endocrine-Related Cancer Endocrine-Related the interaction of ovarian cancer and peritoneal cells using a proteomic approach. We identified several extracellular matrix (ECM) proteins including, fibronectin, TGFBI, periostin, annexin A2 and PAI-1 that were processed as a result of the ovarian cancer– peritoneal cell interaction. This review focuses on the functional role of these proteins in ovarian cancer metastasis. -

Recombinant Rabbit Anti-Human KRT6C Monoclonal Antibody, Clone CQ7176 (CABT- Z232R) This Product Is for Research Use Only and Is Not Intended for Diagnostic Use
Recombinant Rabbit Anti-Human KRT6C Monoclonal Antibody, clone CQ7176 (CABT- Z232R) This product is for research use only and is not intended for diagnostic use. PRODUCT INFORMATION Immunogen Synthetic peptide corresponding to Cytokeratin 6 residues within aa464-564 of Cytokeratin 6 was used as an immunogen. Isotype IgG Source/Host Rabbit Species Reactivity Human Clone CQ7176 Purification ProA affinity purified IgG. Conjugate Unconjugated Applications IHC-P Recommended concentration: HIER; IHC-P: 1:100-1:200 Molecular Weight 60 kDa Cellular Localization Cytoplasm Positive Control Esophagus cancer Format Liquid Concentration Lot specific Size 100 μl Buffer PBS 59%, Sodium azide 0.01%, Glycerol 40%, BSA 0.05%. Preservative 0.01% Sodium azide Storage Store at -20 °C. Avoid freeze/thaw cycles. Ship Wet ice 45-1 Ramsey Road, Shirley, NY 11967, USA Email: [email protected] Tel: 1-631-624-4882 Fax: 1-631-938-8221 1 © Creative Diagnostics All Rights Reserved Warnings This product is for research use only and is not intended for diagnostic use. BACKGROUND Introduction Cytokeratin 6 is an epidermis-specific type I keratin involved in wound healing. Involved in the activation of follicular keratinocytes after wounding, while it does not play a major role in keratinocyte proliferation or migration. This protein always paired with the type-I keratin K16.Cytokeratin 6 have been identified in epidermis only in samples from plantar glabrous skin while in hairy skin this keratin appeared to be absent in interfollicular epidermis but were clearly present in hair follicle outer root sheath, and this protein also consistently expressed in non- keratinizing stratified squamous epithelia.