Broad Transcriptomic Dysregulation Across the Cerebral Cortex in ASD
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Hereditary Spastic Paraplegia: from Genes, Cells and Networks to Novel Pathways for Drug Discovery
brain sciences Review Hereditary Spastic Paraplegia: From Genes, Cells and Networks to Novel Pathways for Drug Discovery Alan Mackay-Sim Griffith Institute for Drug Discovery, Griffith University, Brisbane, QLD 4111, Australia; a.mackay-sim@griffith.edu.au Abstract: Hereditary spastic paraplegia (HSP) is a diverse group of Mendelian genetic disorders affect- ing the upper motor neurons, specifically degeneration of their distal axons in the corticospinal tract. Currently, there are 80 genes or genomic loci (genomic regions for which the causative gene has not been identified) associated with HSP diagnosis. HSP is therefore genetically very heterogeneous. Finding treatments for the HSPs is a daunting task: a rare disease made rarer by so many causative genes and many potential mutations in those genes in individual patients. Personalized medicine through genetic correction may be possible, but impractical as a generalized treatment strategy. The ideal treatments would be small molecules that are effective for people with different causative mutations. This requires identification of disease-associated cell dysfunctions shared across geno- types despite the large number of HSP genes that suggest a wide diversity of molecular and cellular mechanisms. This review highlights the shared dysfunctional phenotypes in patient-derived cells from patients with different causative mutations and uses bioinformatic analyses of the HSP genes to identify novel cell functions as potential targets for future drug treatments for multiple genotypes. Keywords: neurodegeneration; motor neuron disease; spastic paraplegia; endoplasmic reticulum; Citation: Mackay-Sim, A. Hereditary protein-protein interaction network Spastic Paraplegia: From Genes, Cells and Networks to Novel Pathways for Drug Discovery. Brain Sci. 2021, 11, 403. -

A Conserved Amphipathic Helix Is Required for Membrane Tubule Formation by Yop1p
A conserved amphipathic helix is required for PNAS PLUS membrane tubule formation by Yop1p Jacob P. Brady, Jolyon K. Claridge, Peter G. Smith, and Jason R. Schnell1 Department of Biochemistry, University of Oxford, Oxford OX1 3QU, United Kingdom Edited by William F. DeGrado, School of Pharmacy, University of California, San Francisco, CA, and approved December 31, 2014 (received for review August 18, 2014) The integral membrane proteins of the DP1 (deleted in polyposis) C-terminal tubulin binding domains providing a link between ER and reticulon families are responsible for maintaining the high morphology and the cytoskeleton (19). membrane curvature required for both smooth endoplasmic retic- The RHDs are proposed to form hydrophobic hairpins too ulum (ER) tubules and the edges of ER sheets, and mutations in short to traverse the membrane fully, leading to greater dis- these proteins lead to motor neuron diseases, such as hereditary placement of lipids from the outer leaflet than from the in- spastic paraplegia. Reticulon/DP1 proteins contain reticulon ho- ner leaflet. By this mechanism, a combination of hydrophobic mology domains (RHDs) that have unusually long hydrophobic wedging and oligomeric scaffolding could lead to the stabiliza- segments and are proposed to adopt intramembrane helical hair- tion of the tubular ER membrane. However, there is currently pins that stabilize membrane curvature. We have characterized no structural information available for these intramembrane the secondary structure and dynamics of the DP1 family protein domains, and the exact mechanism of curvature generation and produced from the YOP1 gene (Yop1p) and identified a C-termi- stabilization remains unknown. The mechanism for exclusive nal conserved amphipathic helix (APH) that, on its own, interacts localization of RHD proteins to highly curved membranes also strongly with negatively charged membranes and is necessary remains unclear; however, the transmembrane domains have for membrane tubule formation. -

The Transcriptome of Pig Spermatozoa, and Its Role in Fertility
International Journal of Molecular Sciences Article The Transcriptome of Pig Spermatozoa, and Its Role in Fertility Manuel Alvarez-Rodriguez 1,* , Cristina Martinez 1, Dominic Wright 2, Isabel Barranco 3, Jordi Roca 4 and Heriberto Rodriguez-Martinez 1 1 Department of Biomedical & Clinical Sciences (BKV), BKH/Obstetrics & Gynaecology, Faculty of Medicine and Health Sciences, Linköping University, SE-58185 Linköping, Sweden; [email protected] (C.M.); [email protected] (H.R.-M.) 2 Department of Physics, Chemistry and Biology, Faculty of Science and Engineering, Linköping University, SE-58183 Linköping, Sweden; [email protected] 3 Biotechnology of Animal and Human Reproduction (TechnoSperm), Department of Biology, Institute of Food and Agricultural Technology, University of Girona, 17003 Girona, Spain; [email protected] 4 Department of Medicine and Animal Surgery, Faculty of Veterinary Medicine, International Campus for Higher Education and Research “Campus Mare Nostrum”, University of Murcia, 30100 Murcia, Spain; [email protected] * Correspondence: [email protected]; Tel.: +46-(0)729427883 Received: 6 February 2020; Accepted: 24 February 2020; Published: 25 February 2020 Abstract: In the study presented here we identified transcriptomic markers for fertility in the cargo of pig ejaculated spermatozoa using porcine-specific micro-arrays (GeneChip® miRNA 4.0 and GeneChip® Porcine Gene 1.0 ST). We report (i) the relative abundance of the ssc-miR-1285, miR-16, miR-4332, miR-92a, miR-671-5p, miR-4334-5p, miR-425-5p, miR-191, miR-92b-5p and miR-15b miRNAs, and (ii) the presence of 347 up-regulated and 174 down-regulated RNA transcripts in high-fertility breeding boars, based on differences of farrowing rate (FS) and litter size (LS), relative to low-fertility boars in the (Artificial Insemination) AI program. -

Variation in Protein Coding Genes Identifies Information Flow
bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 1 1 2 3 4 5 Variation in protein coding genes identifies information flow as a contributor to 6 animal complexity 7 8 Jack Dean, Daniela Lopes Cardoso and Colin Sharpe* 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Institute of Biological and Biomedical Sciences 25 School of Biological Science 26 University of Portsmouth, 27 Portsmouth, UK 28 PO16 7YH 29 30 * Author for correspondence 31 [email protected] 32 33 Orcid numbers: 34 DLC: 0000-0003-2683-1745 35 CS: 0000-0002-5022-0840 36 37 38 39 40 41 42 43 44 45 46 47 48 49 Abstract bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 2 1 Across the metazoans there is a trend towards greater organismal complexity. How 2 complexity is generated, however, is uncertain. Since C.elegans and humans have 3 approximately the same number of genes, the explanation will depend on how genes are 4 used, rather than their absolute number. -

Hereditary Spastic Paraplegias: Clinical Spectrum in Sudan, Further Deciphering of the Molecular Bases of Autosomal Recessive Forms and New Genes Emerging
Hereditary spastic paraplegias : clinical spectrum in Sudan, further deciphering of the molecular bases of autosomal recessive forms and new genes emerging Liena Elbaghir Omer Elsayed To cite this version: Liena Elbaghir Omer Elsayed. Hereditary spastic paraplegias : clinical spectrum in Sudan, further deciphering of the molecular bases of autosomal recessive forms and new genes emerging. Neurons and Cognition [q-bio.NC]. Université Pierre et Marie Curie - Paris VI; University of Khartoum, 2016. English. NNT : 2016PA066056. tel-01438739 HAL Id: tel-01438739 https://tel.archives-ouvertes.fr/tel-01438739 Submitted on 18 Jan 2017 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Université Pierre et Marie Curie University of Khartoum Cerveau-Cognition-Comportement (ED3C) Institut du Cerveau et de la Moelle Epinière / Equipe Bases Moléculaires, Physiopathologie Et Traitement Des Maladies Neurodégénératives Hereditary spastic paraplegias: clinical spectrum in Sudan, further deciphering of the molecular bases of autosomal recessive forms and new genes emerging -

Copy Number Variation, Chromosome Rearrangement, and Their Association with Recombination During Avian Evolution
Downloaded from genome.cshlp.org on October 1, 2021 - Published by Cold Spring Harbor Laboratory Press Research Copy number variation, chromosome rearrangement, and their association with recombination during avian evolution Martin Vo¨lker,1 Niclas Backstro¨m,2 Benjamin M. Skinner,1,3 Elizabeth J. Langley,1,4 Sydney K. Bunzey,1 Hans Ellegren,2 and Darren K. Griffin1,5 1School of Biosciences, University of Kent, Canterbury, Kent CT2 7NJ, United Kingdom; 2Department of Evolutionary Biology, Evolutionary Biology Centre, Uppsala University, Norbyva¨gen 18D, SE-752 36 Uppsala, Sweden Chromosomal rearrangements and copy number variants (CNVs) play key roles in genome evolution and genetic disease; however, the molecular mechanisms underlying these types of structural genomic variation are not fully understood. The availability of complete genome sequences for two bird species, the chicken and the zebra finch, provides, for the first time, an ideal opportunity to analyze the relationship between structural genomic variation (chromosomal and CNV) and recombination on a genome-wide level. The aims of this study were therefore threefold: (1) to combine bioinformatics, physical mapping to produce comprehensive comparative maps of the genomes of chicken and zebra finch. In so doing, this allowed the identification of evolutionary chromosomal rearrangements distinguishing them. The previously reported interchromosomal conservation of synteny was confirmed, but a larger than expected number of intrachromosomal rearrangements were reported; (2) to hybridize zebra finch genomic DNA to a chicken tiling path microarray and identify CNVs in the zebra finch genome relative to chicken; 32 interspecific CNVs were identified; and (3) to test the hypothesis that there is an association between CNV, chromosomal rearrangements, and recombination by correlating data from (1) and (2) with recombination rate data from a high-resolution genetic linkage map of the zebra finch. -

Agricultural University of Athens
ΓΕΩΠΟΝΙΚΟ ΠΑΝΕΠΙΣΤΗΜΙΟ ΑΘΗΝΩΝ ΣΧΟΛΗ ΕΠΙΣΤΗΜΩΝ ΤΩΝ ΖΩΩΝ ΤΜΗΜΑ ΕΠΙΣΤΗΜΗΣ ΖΩΙΚΗΣ ΠΑΡΑΓΩΓΗΣ ΕΡΓΑΣΤΗΡΙΟ ΓΕΝΙΚΗΣ ΚΑΙ ΕΙΔΙΚΗΣ ΖΩΟΤΕΧΝΙΑΣ ΔΙΔΑΚΤΟΡΙΚΗ ΔΙΑΤΡΙΒΗ Εντοπισμός γονιδιωματικών περιοχών και δικτύων γονιδίων που επηρεάζουν παραγωγικές και αναπαραγωγικές ιδιότητες σε πληθυσμούς κρεοπαραγωγικών ορνιθίων ΕΙΡΗΝΗ Κ. ΤΑΡΣΑΝΗ ΕΠΙΒΛΕΠΩΝ ΚΑΘΗΓΗΤΗΣ: ΑΝΤΩΝΙΟΣ ΚΟΜΙΝΑΚΗΣ ΑΘΗΝΑ 2020 ΔΙΔΑΚΤΟΡΙΚΗ ΔΙΑΤΡΙΒΗ Εντοπισμός γονιδιωματικών περιοχών και δικτύων γονιδίων που επηρεάζουν παραγωγικές και αναπαραγωγικές ιδιότητες σε πληθυσμούς κρεοπαραγωγικών ορνιθίων Genome-wide association analysis and gene network analysis for (re)production traits in commercial broilers ΕΙΡΗΝΗ Κ. ΤΑΡΣΑΝΗ ΕΠΙΒΛΕΠΩΝ ΚΑΘΗΓΗΤΗΣ: ΑΝΤΩΝΙΟΣ ΚΟΜΙΝΑΚΗΣ Τριμελής Επιτροπή: Aντώνιος Κομινάκης (Αν. Καθ. ΓΠΑ) Ανδρέας Κράνης (Eρευν. B, Παν. Εδιμβούργου) Αριάδνη Χάγερ (Επ. Καθ. ΓΠΑ) Επταμελής εξεταστική επιτροπή: Aντώνιος Κομινάκης (Αν. Καθ. ΓΠΑ) Ανδρέας Κράνης (Eρευν. B, Παν. Εδιμβούργου) Αριάδνη Χάγερ (Επ. Καθ. ΓΠΑ) Πηνελόπη Μπεμπέλη (Καθ. ΓΠΑ) Δημήτριος Βλαχάκης (Επ. Καθ. ΓΠΑ) Ευάγγελος Ζωίδης (Επ.Καθ. ΓΠΑ) Γεώργιος Θεοδώρου (Επ.Καθ. ΓΠΑ) 2 Εντοπισμός γονιδιωματικών περιοχών και δικτύων γονιδίων που επηρεάζουν παραγωγικές και αναπαραγωγικές ιδιότητες σε πληθυσμούς κρεοπαραγωγικών ορνιθίων Περίληψη Σκοπός της παρούσας διδακτορικής διατριβής ήταν ο εντοπισμός γενετικών δεικτών και υποψηφίων γονιδίων που εμπλέκονται στο γενετικό έλεγχο δύο τυπικών πολυγονιδιακών ιδιοτήτων σε κρεοπαραγωγικά ορνίθια. Μία ιδιότητα σχετίζεται με την ανάπτυξη (σωματικό βάρος στις 35 ημέρες, ΣΒ) και η άλλη με την αναπαραγωγική -

Loss of Association of REEP2 with Membranes Leads to Hereditary Spastic Paraplegia
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector REPORT Loss of Association of REEP2 with Membranes Leads to Hereditary Spastic Paraplegia Typhaine Esteves,1,2,3,4 Alexandra Durr,1,2,3,5 Emeline Mundwiller,6 Jose´ L. Loureiro,7 Maxime Boutry,1,2,3 Michael A. Gonzalez,8 Julie Gauthier,9,12 Khalid H. El-Hachimi,1,2,3,4 Christel Depienne,1,2,3,5 Marie-Paule Muriel,1,2,3 Rafael F. Acosta Lebrigio,8 Marion Gaussen,1,2,3,4 Anne Noreau,9 Fiorella Speziani,8 Alexandre Dionne-Laporte,9 Jean-Franc¸ois Deleuze,10 Patrick Dion,9 Paula Coutinho,7 Guy A. Rouleau,9 Stephan Zuchner,8 Alexis Brice,1,2,3,5,6 Giovanni Stevanin,1,2,3,4,6,11,* and Fre´de´ric Darios1,2,3,11,* Hereditary spastic paraplegias (HSPs) are clinically and genetically heterogeneous neurological conditions. Their main pathogenic mech- anisms are thought to involve alterations in endomembrane trafficking, mitochondrial function, and lipid metabolism. With a combi- nation of whole-genome mapping and exome sequencing, we identified three mutations in REEP2 in two families with HSP: a missense variant (c.107T>A [p.Val36Glu]) that segregated in the heterozygous state in a family with autosomal-dominant inheritance and a missense change (c.215T>A [p.Phe72Tyr]) that segregated in trans with a splice site mutation (c.105þ3G>T) in a family with auto- somal-recessive transmission. REEP2 belongs to a family of proteins that shape the endoplasmic reticulum, an organelle that was altered in fibroblasts from an affected subject. -

Massive Sequencing of 70 Genes Reveals a Myriad of Missing Genes Or Mechanisms to Be Uncovered in Hereditary Spastic Paraplegias
European Journal of Human Genetics (2017) 25, 1217–1228 Official journal of The European Society of Human Genetics www.nature.com/ejhg ARTICLE Massive sequencing of 70 genes reveals a myriad of missing genes or mechanisms to be uncovered in hereditary spastic paraplegias Sara Morais1,2,3,4,5,6,7,8, Laure Raymond4,5,6,7,8, Mathilde Mairey4,5,6,7,8, Paula Coutinho1,2, Eva Brandão9, Paula Ribeiro9, José Leal Loureiro1,2,9, Jorge Sequeiros1,2,3, Alexis Brice4,5,6,7,10, Isabel Alonso*,1,2,3,11 and Giovanni Stevanin4,5,6,7,8,10,11 Hereditary spastic paraplegias (HSP) are neurodegenerative disorders characterized by lower limb spasticity and weakness that can be complicated by other neurological or non-neurological signs. Despite a high genetic heterogeneity (460 causative genes), 40–70% of the families remain without a molecular diagnosis. Analysis of one of the pioneer cohorts of 193 HSP families generated in the early 1990s in Portugal highlighted that SPAST and SPG11 are the most frequent diagnoses. We have now explored 98 unsolved families from this series using custom next generation sequencing panels analyzing up to 70 candidate HSP genes. We identified the likely disease-causing variant in 20 of the 98 families with KIF5A being the most frequently mutated gene. We also found 52 variants of unknown significance (VUS) in 38% of the cases. These new diagnoses resulted in 42% of solved cases in the full Portuguese cohort (81/193). Segregation of the variants was not always compatible with the presumed inheritance, indicating that the analysis of all HSP genes regardless of the inheritance mode can help to explain some cases. -
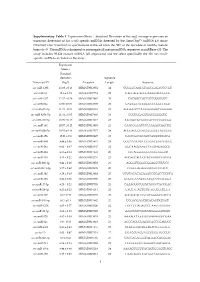
Supplementary Table 1. Expression
Supplementary Table 1. Expression (Mean Standard Deviation of the log2 average expression or transcript detection) of Sus scrofa specific miRNAs detected by the GeneChip™ miRNA 4.0 Array (ThermoFisher Scientific) in spermatozoa retrieved from the SRF of the ejaculate of healthy mature boars (n=3). The miRNA is designed to interrogate all mature miRNA sequences in miRBase v20. The array includes 30.424 mature miRNA (all organisms) and we select specifically the 326 Sus scrofa- specific miRNAs included in the array. Expression Mean ± Standard Deviation Sequence Transcript ID (log2) Accession Length Sequence ssc-miR-1285 13.98 ± 0.13 MIMAT0013954 24 CUGGGCAACAUAGCGAGACCCCGU ssc-miR-16 12.6 ± 0.74 MIMAT0007754 22 UAGCAGCACGUAAAUAUUGGCG ssc-miR-4332 12.32 ± 0.29 MIMAT0017962 20 CACGGCCGCCGCCGGGCGCC ssc-miR-92a 12.06 ± 0.09 MIMAT0013908 22 UAUUGCACUUGUCCCGGCCUGU ssc-miR-671-5p 11.73 ± 0.54 MIMAT0025381 24 AGGAAGCCCUGGAGGGGCUGGAGG ssc-miR-4334-5p 11.31 ± 0.05 MIMAT0017966 19 CCCUGGAGUGACGGGGGUG ssc-miR-425-5p 10.99 ± 0.15 MIMAT0013917 23 AAUGACACGAUCACUCCCGUUGA ssc-miR-191 10.57 ± 0.22 MIMAT0013876 23 CAACGGAAUCCCAAAAGCAGCUG ssc-miR-92b-5p 10.53 ± 0.18 MIMAT0017377 24 AGGGACGGGACGCGGUGCAGUGUU ssc-miR-15b 10.01 ± 0.9 MIMAT0002125 22 UAGCAGCACAUCAUGGUUUACA ssc-miR-30d 9.89 ± 0.36 MIMAT0013871 24 UGUAAACAUCCCCGACUGGAAGCU ssc-miR-26a 9.62 ± 0.47 MIMAT0002135 22 UUCAAGUAAUCCAGGAUAGGCU ssc-miR-484 9.55 ± 0.14 MIMAT0017974 20 CCCAGGGGGCGACCCAGGCU ssc-miR-103 9.53 ± 0.22 MIMAT0002154 23 AGCAGCAUUGUACAGGGCUAUGA ssc-miR-296-3p 9.41 ± 0.26 MIMAT0022958 -

Mitotic Checkpoints and Chromosome Instability Are Strong Predictors of Clinical Outcome in Gastrointestinal Stromal Tumors
MITOTIC CHECKPOINTS AND CHROMOSOME INSTABILITY ARE STRONG PREDICTORS OF CLINICAL OUTCOME IN GASTROINTESTINAL STROMAL TUMORS. Pauline Lagarde1,2, Gaëlle Pérot1, Audrey Kauffmann3, Céline Brulard1, Valérie Dapremont2, Isabelle Hostein2, Agnès Neuville1,2, Agnieszka Wozniak4, Raf Sciot5, Patrick Schöffski4, Alain Aurias1,6, Jean-Michel Coindre1,2,7 Maria Debiec-Rychter8, Frédéric Chibon1,2. Supplemental data NM cases deletion frequency. frequency. deletion NM cases Mand between difference the highest setswith of theprobe a view isdetailed panel Bottom frequently. sorted totheless deleted theprobe are frequently from more and thefrequency deletion represent Yaxes inblue. are cases (NM) metastatic for non- frequencies Corresponding inmetastatic (red). probe (M)cases sets figureSupplementary 1: 100 100 20 40 60 80 20 40 60 80 0 0 chr14 1 chr14 88 chr14 175 chr14 262 chr9 -MTAP 349 chr9 -MTAP 436 523 chr9-CDKN2A 610 Histogram presenting the 2000 more frequently deleted deleted frequently the 2000 more presenting Histogram chr9-CDKN2A 697 chr9-CDKN2A 784 chr9-CDKN2B 871 chr9-CDKN2B 958 chr9-CDKN2B 1045 chr22 1132 chr22 1219 chr22 1306 chr22 1393 1480 1567 M NM 1654 1741 1828 1915 M NM GIST14 GIST2 GIST16 GIST3 GIST19 GIST63 GIST9 GIST38 GIST61 GIST39 GIST56 GIST37 GIST47 GIST58 GIST28 GIST5 GIST17 GIST57 GIST47 GIST58 GIST28 GIST5 GIST17 GIST57 CDKN2A Supplementary figure 2: Chromosome 9 genomic profiles of the 18 metastatic GISTs (upper panel). Deletions and gains are indicated in green and red, respectively; and color intensity is proportional to copy number changes. A detailed view is given (bottom panel) for the 6 cases presenting a homozygous 9p21 deletion targeting CDKN2A locus (dark green).