Investigation of Layered Calcium Phosphates and Related Materials for Biomaterial Applications
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

How to Fortify Beverages with Calcium by Dr
Ingredients How to Fortify Beverages With Calcium by Dr. Gerhard Gerstner Along with current developments of the overall functional foods market, the use of minerals and especially calcium salts is expected to exhibit strong growth rates. In contrary to other functional ingredients, calcium is widely known as being beneficial for human health and there is no need to explain its nutritional ad- vantages to the customer. According to Leatherhead International, future trends include growing consumer concern regarding osteoporosis and bone health, leading to increased sales of calcium salts. The con- observation is seen as being one of tinuous market growth drives mineral the main factors causing osteo- Common calium sources salt suppliers to offer not only one porosis 2 .As a consequence, national for beverage fortification product but rather a range of different authorities all over the world have calcium salts and granulations to be recently reconsidered recommend- Table 1 shows a typical range of able to tune them to industrial cus- ations in order to take remedial calcium fortified beverages which tomers’ applications. This article measures against calcium deficiency have been seen in European and US discusses important nutritional, and accordingly, to reduce the risk of supermarkets recently. Practically technological as well as economical osteoporosis. In this respect, the US every type of beverage such as aspects of calcium in beverages with National Institute of Health (NIH) has mineral water, soy milk, energy drink, a focus on our company’s products increased the amounts of optimal nectar or juice does have a fortified Tricalcium Citrate, Calcium Gluconate daily calcium intake and defined product line already. -

Tricalcium Phosphate Is Inappropriate As a Universal Selection Factor for Isolating and Testing Phosphate-Solubilizing Bacteria
Biol Fertil Soils (2013) 49:465–479 DOI 10.1007/s00374-012-0737-7 ORIGINAL PAPER Tricalcium phosphate is inappropriate as a universal selection factor for isolating and testing phosphate-solubilizing bacteria that enhance plant growth: a proposal for an alternative procedure Yoav Bashan & Alexander A. Kamnev & Luz E. de-Bashan Received: 26 June 2012 /Revised: 16 August 2012 /Accepted: 30 August 2012 /Published online: 2 October 2012 # Springer-Verlag 2012 Abstract Literature analysis and chemical considerations Other compounds are also tested, but on a very small of biological phosphate solubilization have shown that the scale. These phosphates (P), mainly Fe-P, Al-P, and commonlyusedselectionfactor for this trait, tricalcium several Ca-P, are even less soluble than TCP in water. phosphate (TCP), is relatively weak and unreliable as a Because soils greatly vary by pH and several chemical universal selection factor for isolating and testing considerations, it appears that there is no metal-P com- phosphate-solubilizing bacteria (PSB) for enhancing plant pound that can serve as the universal selection factor for growth. Most publications describing isolation of PSB PSB. A practical approach is to use a combination of employed TCP. The use of TCP usually yields many (up two or three metal-P compounds together or in tandem, to several thousands per study) isolates “supposedly” PSB. according to the end use of these bacteria—Ca-P com- When these isolates are further tested for direct contribution pounds (including rock phosphates) for alkaline soils, of phosphorus to the plants, only a very few are true PSB. Fe-P and Al-P compounds for acidic soils, and phytates for soils rich in organic P. -

Brochure-Product-Range.Pdf
PRODUCT RANGE 2015 edition ANSI Standard 60 NSF® CERTIFIED HALAL M ISLAMIC FOOD AND NUTRITION ® COUNCIL OF AMERICA Rue Joseph Wauters, 144 ISO 9001:2008 (Quality) / OHSAS 18001:2007 (Health/ B-4480 Engis Safety) / ISO 14001:2004 (Environment) / ISO 22000:2005 www.globulebleu.com (Food Safety) / FSSC 22000:2013 (Food Safety). Tel. +32 (0) 4 273 93 58 Our food grade phosphates are allergen free, GMO free, Fax. +32 (0) 4 275 68 36 BSE/TSE free. www.prayon.com mail. [email protected] Design by www.prayon.com PRODUCT RANGE | 11 TABLE OF CONTENTS HORTICULTURE APPLICATIONS HORTIPRAY® RANGE FOR HORTICULTURE* FOOD AND INDUSTRIAL APPLICATIONS PRODUCT NAME Bulk density P O pH N-NH Made 2 5 4 MONOAMMONIUM PHOSPHATE - NH4H2PO4 in 3 3 % 1% % Sodium orthophosphates ................................................................................... 03 g/cm lbs/ft indicative indicative indicative Water-soluble fertilisers. Sodium pyrophosphates .................................................................................... 04 HORTIPRAY® MAP Horticultural Grade 0.9 56 61 4.5 12 Sodium tripolyphosphates ................................................................................. 05 HORTIPRAY® MAP 12.60 Horticultural Grade 0.9 56 60 5 12.1 Water-soluble fertilisers; Sodium polyphosphates ..................................................................................... 06 HORTIPRAY® MAP anticalc Horticultural Grade 0.9 56 61 4.5 12 preventive action against clogging. Potassium orthophosphates ............................................................................. -

Tricalcium Phosphate: an X-Ray Diffraction, Solid State NMR and ATR-FTIR Study
Journal of Functional Biomaterials Article Strontium and Zinc Substitution in β-Tricalcium Phosphate: An X-ray Diffraction, Solid State NMR and ATR-FTIR Study Elisa Boanini 1 , Massimo Gazzano 2,* , Carlo Nervi 3 , Michele R. Chierotti 3 , Katia Rubini 1, Roberto Gobetto 3 and Adriana Bigi 1 1 Department of Chemistry “Giacomo Ciamician”, Alma Mater Studiorum-University of Bologna, 40126 Bologna, Italy; [email protected] (E.B.); [email protected] (K.R.); [email protected] (A.B.) 2 ISOF-CNR, via Gobetti 101, 40129 Bologna, Italy 3 Department of Chemistry, University of Torino, via P. Giuria 7, 10125 Torino, Italy; [email protected] (C.N.); [email protected] (M.R.C.); [email protected] (R.G.) * Correspondence: [email protected]; Tel.: +39-051-2099552 Received: 27 March 2019; Accepted: 28 April 2019; Published: 5 May 2019 Abstract: β-tricalcium phosphate (β-TCP) is one of the most common bioceramics, widely applied in bone cements and implants. Herein we synthesized β-TCP by solid state reaction in the presence of increasing amounts of two biologically active ions, namely strontium and zinc, in order to clarify the structural modifications induced by ionic substitution. The results of X-ray diffraction analysis indicate that zinc can substitute for calcium into a β-TCP structure up to about 10 at% inducing a reduction of the cell parameters, whereas the substitution occurs up to about 80 at% in the case of strontium, which provokes a linear increase of the lattice constants, and a slight modification into a more symmetric structure. -
Material Safety Data Sheet
Material safety data sheet Product name: MSDS date created:Vitoss ™ Scaffold Manufacturer16 June 2021 Australian supplier New Zealand supplier Name: Address: DSM Biomedical Stryker Australia Stryker New Zealand 735 Pennsylvania Drive 8 Herbert St 511 Mt Wellington Highway Phone No: Exton, PA 19341 U.S.A St Leonards NSW Auckland, New Zealand 1060 Fax No: Australia 2065 +484 713-2100 +61 02 9467 1000 +64 09 573 1890 - +61 02 9467 1010 +64 09 573 1891 VITOSS™ SCAFFOLD 1. IDENTIFICATION OF THE MATERIAL AND SUPPLIER Product Name Vitoss™ Scaffold Synonym(s) BETA-TCP • CALCIUM PHOSPHATE • CALCIUM PHOSPHATE TRIBASIC • TCP • VITOSS SCAFFOLD Use(s) ORTHOPAEDIC APPLICATIONS Manufacturer Australian Supplier New Zealand Supplier Name: DSM Biomedical Stryker Australia Stryker New Zealand Address: 735 Pennsylvania Drive 8 Herbert St, 511 Mt Wellington Highway, Exton, PA 19341 U.S.A St Leonards, NSW, Auckland, Australia, 2065 New Zealand, 1060 Phone No: +484 713-2100 +61 02 9467 1000 +64 09 573 1890 Fax No: - +61 02 9467 1010 +64 09 573 1891 EMERGENCY - 13 11 26 0800 764 766 Contact Person: Nikin Desai, [email protected] 2. HAZARDS IDENTIFICATION CLASSIFIED AS HAZARDOUS ACCORDING TO HAZARDOUS SUBSTANCES [CLASSIFICATION] REGULATIONS 2001. NOT CLASSIFIED AS HAZARDOUS ACCORDING TO THE SAFEWORK AUSTRALIA CRITERIA. HSNO CLASSIFICATION 6.1E Substances that are acutely toxic. 9.1D Substances that are slightly harmful to the aquatic environment or are otherwise designed for biocidal action. HAZARD STATEMENT H303 May be harmful if swallowed. H401 Toxic to aquatic life. H402 Harmful to aquatic life. H413 May cause long lasting harmful effects to aquatic life. -

Shelf Life and Storage Conditions Statement Icl
Webster Groves Technical Center 373 Marshall Avenue Webster Groves, MO 63119 1-855-ICL-SPEC (1-855-425-7732) www.icl-pp.com SHELF LIFE AND STORAGE CONDITIONS STATEMENT ICL Performance Products, ICL Food Specialties (a division of ICL Performance Products), and ICL Fosfatos y Aditivos México S.A. de C.V. manufacture and/or market high quality phosphates, phosphoric acid, sulfates, adipic acid, sea salt, licorice root extract products, and specialty food ingredient blends for use in food, pharmaceutical, and/or industrial applications. The recommended storage conditions are for materials to be kept at ambient temperatures, low to moderate humidity and in the original packaging. Materials stacked during long-term storage may develop some pressure compaction and require screening or milling before use. This is particularly true of powdered products. In addition, Licresse™ products must be stored in sealed packaging that prevents exposure to light, and under cool and dry conditions. ICL does not guarantee the performance of our products indefinitely since we have no control over how materials are stored. However, if the recommended storage conditions are maintained, performance should be assured for the shelf life listed in the attached table, and in the case of some products, for a much longer period of time. ICL will not recertify expired products for shelf life extension. Lori L. Klopf, Ph.D. Regulatory Affairs Manager Food & Pharmaceutical Ingredients ICL Food Specialties (a division of ICL Performance Products LP) February 5, 2015 The information stated herein is presented in good faith and is believed to be correct as of the date specified in this statement. -

Drug Registration Guidance Document
NATIONAL PHARMACEUTICAL CONTROL BUREAU MINISTRY OF HEALTH MALAYSIA PETALING JAYA DRUG REGISTRATION GUIDANCE DOCUMENT PREAMBLE This “DRUG REGISTRATION GUIDANCE DOCUMENT” will serve as the reference guide for both pharmaceutical products for human use and traditional products. It will replace the “Guidelines for Application for Registration of Pharmaceutical Products” Third Edition of October 1993 and “Garispanduan Permohonan Pendaftaran Keluaran Ubat Tradisional” Second Edition, December 1998. The contents of this version include: Updated information relating to administrative requirements and procedures. Information on Drug Control Authority (DCA) policies currently applicable. Guidelines on the on-line application process and requirements which will incorporate the ASEAN technical requirements and standards for pharmaceuticals (where applicable). An on-going review of policy matters will continue, taking into account the global regulatory environment, to allow for timely and pertinent changes. Information relating to DCA policy decisions is current up to its 220th meeting on 01 Oktober 2009. Please visit the National Pharmaceutical Control Bureau (NPCB) website at http://www.bpfk.gov.my for updates in regulatory information. March 2010 Revision 1 __________________________________________________________________________________ DRUG REGISTRATION GUIDANCE DOCUMENT (MALAYSIA) List of amendments / changes: March 2010 Revision 6. REGULATORY OUTCOME - Addition of sub-point 6.7 Reapplication of Rejected Products Section D: Label (Mockup) For Immediate Container, Outer Carton and Proposed Package Insert - Addition of phrase ‘Product that contains Nevirapine: Addition of phrase “Retriction of Nevirapine use in patient with CD4+cell count greater than 250cells/mm3 on the product label.Product Holder is fully responsible to inform prescriber pertaining to the restriction in use of Nevirapine. Updated on 30 March 2010 from DCA Policy 3/2009 13.8 Guide for Implementation of Patient Dispensing Pack for Pharmaceutical Products in Malaysia. -

Synthesis and Characterization of Powders Calcium Phosphate for Biomedical Applications
Seventh International Latin American Conference on Powder Technology, November 08-10, Atibaia, SP, Brazil SYNTHESIS AND CHARACTERIZATION OF POWDERS CALCIUM PHOSPHATE FOR BIOMEDICAL APPLICATIONS De Oliveira, D.M.P.1,a, Prants W.T.2,b, Camargo, N.H.A.3,c, Gemelli, E4,d. 1,2,3,4Universidade do Estado de Santa Catarina (UDESC), Centro de Ciências Tecnológicas (CCT), Campus Universitário Prof. Avelino Marcante S/N - Bairro Bom Retiro – Joinville - SC – Brasil CEP: 89223 - 100 - CX. Postal 631 - Fone (0xx47) 431- 7200 - Fax (0xx47) 431-7240 e-mail: [email protected] , [email protected], [email protected], [email protected]. Keywords: Phosphate Calcium, Thermal Treatments, Characterization. Abstract. Scientists of different areas research the bioceramics as new materials to substitute parts of the human body. The bioceramics of the calcium phosphate have the advantage present similar chemical composition to the structure of the bony apatite of the human skeleton. In this study, calcium phosphate powder was synthesized chemically using the solution of phosphorus pentoxide (P2O5) and calcium oxide (CaO) necessary for molar Ca/P =1.67. These works aim the study of different thermal treatments, physics and of the microstructure properties. For characterization the bony matrix were used the techniques of: X-ray diffraction (DRX); Scanning Electronic Microscopy (SEM) and Differential Scanning Calorimetry (DSC). 1. INTRODUCION Calcium phosphate based bioceramics have received much attention as bone substitutes due to their chemical similarity to natural bone, their bioactivity and their biocompatibility. This class of biomaterials are promising applications in orthopedics, traumatology and ondontology [1]. These bioceramics may be used for attachment of implants in reconstruction of bone tissue and the controlled release of drugs in treatments ontological. -

Simple Recycling of Biowaste Eggshells to Various Calcium
www.nature.com/scientificreports OPEN Simple recycling of biowaste eggshells to various calcium phosphates for specifc industries Nongnuch Laohavisuti1, Banjong Boonchom2,3*, Wimonmat Boonmee4, Kittichai Chaiseeda5* & Somkiat Seesanong6 Egg consumption is very high throughout the world and with it comes enormous amount of waste eggshells. To reduce and utilize these wastes, eggshell wastes were simply transformed to low- or high-purity calcium carbonate grades by washing, crushing, and drying to use as raw materials for producing highly valuable calcium phosphate products. Low-purity calcium carbonate grade was used to prepare triple superphosphate for using in fertilizer industry, whereas high-purity calcium carbonate grade was used to produce dicalcium phosphate dihydrate, monocalcium phosphate monohydrate, and tricalcium phosphate for using in mineral feed and food additive industries. All calcium phosphate samples obtained by simple, rapid, cheap, and environmentally safe method using eggshells and phosphoric acid were identifed and their structural phases and impurities were determined by XRF, XRD and FTIR techniques. Thermal behaviors of raw materials and the prepared calcium phosphates excepted tricalcium phosphate were investigated by TG/DTG techniques. The methodologies described here will be useful to manage eggshells by converting them to highly valuable products, which can solve eggshell wastes problem from industries and communities. This fnding supports the viewpoint of zero waste operation to produce value-added products for obtaining sustainable development, which may be selected as an alternative way for material recycling and waste management in the future. Agro-food from felds of industries and communities produce a substantial amount of pollution every day, so it is becoming urgent to manage this problem1. -

A Review on Calcium Pyrophosphate and Other Related Phosphate Nano Bio-Materials and Their Applications
4R4ev. Adv. Mater. Sci. 48 (2017) 44-57 S. R. Vasant and M. J. Joshi A REVIEW ON CALCIUM PYROPHOSPHATE AND OTHER RELATED PHOSPHATE NANO BIO-MATERIALS AND THEIR APPLICATIONS Sonal R. Vasant1 and M. J. Joshi2 1Government Engineering College, Rajkot, Gujarat, India 2Department of Physics, Saurashtra University, Rajkot, Gujarat, India Received: July 29, 2016 Abstract. Over the past few decades, a little word with big potential has been rapidly insinuating into the world’s consciousness, nano. The nano-scale structures permit the control of funda- mental properties of the materials without changing their chemical structure. Living organisms can create amazing ways to produce high performance materials that involve not only creation of implants, replacing bone tissues and organs, but also synthesis of biologically active materials promoting the fullest restoration of tissues and maintenance of necessary functions of an organ- ism. In the biomaterial research field, nowadays, a great attention is given to bone substitute materials, particularly, calcium phosphate based bio-materials play important roles in clinical applications as it possesses high bio-compatibility and excellent ability to undergo varying de- grees of resorbability. These materials show a positive interaction with living tissue that includes differentiation of immature cells towards bone cells and also have chemical bonding to the bone along the interface, thought to be triggered by the adsorption of bone growth-mediating proteins at the bio-materials surface. In the present review paper an attempt is made to cover properties and applications of various phosphate nano bio-materials like calcium phosphate, calcium polyphosphate, calcium pyrophosphate etc over a time span right from 1920 to 2016. -
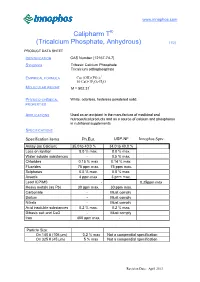
Tricalcium Phosphate, Anhydrous) (1/2)
www.innophos.com ® Calipharm T (Tricalcium Phosphate, Anhydrous) (1/2) PRODUCT DATA SHEET IDENTIFICATION CAS Number [12167-74-7] Tribasic Calcium Phosphate SYNONYM Tricalcium orthophosphate * EMPIRICAL FORMULA Ca5 (OH) (PO4)3 10 CaO•3P2O5•H2O OLECULAR WEIGHT * M Mr = 502.31 PHYSICO-CHEMICAL White, odorless, tasteless powdered solid. PROPERTIES APPLICATIONS Used as an excipient in the manufacture of medicinal and nutraceutical products and as a source of calcium and phosphorus in nutritional supplements. SPECIFICATIONS Specification items Ph.Eur. USP-NF Innophos Spec. Assay (as Calcium) 35.0 to 40.0 % 34.0 to 40.0 % Loss on Ignition 8.0 % max. 8.0 % max. Water soluble substances - 0.5 % max. Chlorides 0.15 % max 0.14 % max. Fluorides 75 ppm max. 75 ppm max. Sulphates 0.5 % max. 0.8 % max. Arsenic 4 ppm max 3 ppm max. Lead ICP/MS - - 0.25ppm max Heavy metals (as Pb) 30 ppm max. 30 ppm max. Carbonate - Must comply Barium - Must comply Nitrate - Must comply Acid insoluble substances 0.2 % max. 0.2 % max. Dibasic salt and CaO - Must comply Iron 400 ppm max. - Particle Size: On 140 # (106 µm) 0.2 % max Not a compendial specification On 325 # (45 µm) 5 % max Not a compendial specification Revision Date: April 2013 www.innophos.com ® Calipharm T (Tricalcium Phosphate, Anhydrous) (2/2) TYPICAL PROPERTIES pH (20 % slurry) 6.8 Tapped density 0.56 g/ml (35 lbs/ft3 ) Calcium content (unignited basis) 37.6 % Phosphorus content (unignited basis) 17.3 % Particle size: on 140 # ( 106 µm) 0.01 % on 325 # ( 45 µm) 1 % REGULATORY / QUALITY This product meets the requirements of the NF/EP compendial specifications, current edition. -

Efficacy of Fluoride Varnishes for Preventing Enamel Demineralization After Interproximal Enamel Reduction
RESEARCH ARTICLE Efficacy of fluoride varnishes for preventing enamel demineralization after interproximal enamel reduction. Qualitative and quantitative evaluation Ascensio n Vicente1☯*, Antonio Jose Ortiz Ruiz2☯, BeleÂn Manuela GonzaÂlez Paz1³, Jose GarcõÂa Lo pez1³, Luis-Alberto Bravo-GonzaÂlez1☯ 1 Department of Orthodontics, Faculty of Medicine, University of Murcia, Hospital Morales Meseguer, 2ã planta, C/ MarqueÂs de los VeÂlez s/n, Murcia, Spain, 2 Department of Integral Pediatric Dentistry, Faculty of Medicine, University of Murcia, Hospital Morales Meseguer, 2ã planta, C/ MarqueÂs de los VeÂlez s/n, Murcia, Spain a1111111111 a1111111111 ☯ These authors contributed equally to this work. a1111111111 ³ These authors also contributed equally to this work. * [email protected] a1111111111 a1111111111 Abstract OPEN ACCESS Citation: Vicente A, Ortiz Ruiz AJ, GonzaÂlez Paz Objectives BM, GarcõÂa LoÂpez J, Bravo-GonzaÂlez L-A (2017) To evaluate quantitatively and qualitatively the changes produced to enamel after interproxi- Efficacy of fluoride varnishes for preventing enamel mal reduction and subjected to demineralization cycles, after applying a fluoride varnish demineralization after interproximal enamel reduction. Qualitative and quantitative evaluation. (Profluorid) and a fluoride varnish containing tricalcium phosphate modified by fumaric acid PLoS ONE 12(4): e0176389. https://doi.org/ (Clinpro White). 10.1371/journal.pone.0176389 Editor: Giuseppe Remuzzi, Istituto Di Ricerche Farmacologiche Mario Negri, ITALY Materials and methods Received: October 6, 2016 138 interproximal dental surfaces were divided into six groups: 1) Intact enamel; 2) Intact Accepted: April 10, 2017 enamel + demineralization cycles (DC); 3) Interproximal Reduction (IR); 4) IR + DC; 5) IR + Published: April 21, 2017 Profluorid + DC; 6) IR + Clinpro White + DC. IR was performed with a 0.5 mm cylindrical dia- mond bur.