ARF4 Signalling Pathway Mediates the Response to Golgi Stress and Susceptibility to Pathogens
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 Supporting Information for a Microrna Network Regulates
Supporting Information for A microRNA Network Regulates Expression and Biosynthesis of CFTR and CFTR-ΔF508 Shyam Ramachandrana,b, Philip H. Karpc, Peng Jiangc, Lynda S. Ostedgaardc, Amy E. Walza, John T. Fishere, Shaf Keshavjeeh, Kim A. Lennoxi, Ashley M. Jacobii, Scott D. Rosei, Mark A. Behlkei, Michael J. Welshb,c,d,g, Yi Xingb,c,f, Paul B. McCray Jr.a,b,c Author Affiliations: Department of Pediatricsa, Interdisciplinary Program in Geneticsb, Departments of Internal Medicinec, Molecular Physiology and Biophysicsd, Anatomy and Cell Biologye, Biomedical Engineeringf, Howard Hughes Medical Instituteg, Carver College of Medicine, University of Iowa, Iowa City, IA-52242 Division of Thoracic Surgeryh, Toronto General Hospital, University Health Network, University of Toronto, Toronto, Canada-M5G 2C4 Integrated DNA Technologiesi, Coralville, IA-52241 To whom correspondence should be addressed: Email: [email protected] (M.J.W.); yi- [email protected] (Y.X.); Email: [email protected] (P.B.M.) This PDF file includes: Materials and Methods References Fig. S1. miR-138 regulates SIN3A in a dose-dependent and site-specific manner. Fig. S2. miR-138 regulates endogenous SIN3A protein expression. Fig. S3. miR-138 regulates endogenous CFTR protein expression in Calu-3 cells. Fig. S4. miR-138 regulates endogenous CFTR protein expression in primary human airway epithelia. Fig. S5. miR-138 regulates CFTR expression in HeLa cells. Fig. S6. miR-138 regulates CFTR expression in HEK293T cells. Fig. S7. HeLa cells exhibit CFTR channel activity. Fig. S8. miR-138 improves CFTR processing. Fig. S9. miR-138 improves CFTR-ΔF508 processing. Fig. S10. SIN3A inhibition yields partial rescue of Cl- transport in CF epithelia. -

Novel Driver Strength Index Highlights Important Cancer Genes in TCGA Pancanatlas Patients
medRxiv preprint doi: https://doi.org/10.1101/2021.08.01.21261447; this version posted August 5, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . Novel Driver Strength Index highlights important cancer genes in TCGA PanCanAtlas patients Aleksey V. Belikov*, Danila V. Otnyukov, Alexey D. Vyatkin and Sergey V. Leonov Laboratory of Innovative Medicine, School of Biological and Medical Physics, Moscow Institute of Physics and Technology, 141701 Dolgoprudny, Moscow Region, Russia *Corresponding author: [email protected] NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. 1 medRxiv preprint doi: https://doi.org/10.1101/2021.08.01.21261447; this version posted August 5, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . Abstract Elucidating crucial driver genes is paramount for understanding the cancer origins and mechanisms of progression, as well as selecting targets for molecular therapy. Cancer genes are usually ranked by the frequency of mutation, which, however, does not necessarily reflect their driver strength. Here we hypothesize that driver strength is higher for genes that are preferentially mutated in patients with few driver mutations overall, because these few mutations should be strong enough to initiate cancer. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

The Database of Chromosome Imbalance Regions and Genes
Lo et al. BMC Cancer 2012, 12:235 http://www.biomedcentral.com/1471-2407/12/235 RESEARCH ARTICLE Open Access The database of chromosome imbalance regions and genes resided in lung cancer from Asian and Caucasian identified by array-comparative genomic hybridization Fang-Yi Lo1, Jer-Wei Chang1, I-Shou Chang2, Yann-Jang Chen3, Han-Shui Hsu4, Shiu-Feng Kathy Huang5, Fang-Yu Tsai2, Shih Sheng Jiang2, Rajani Kanteti6, Suvobroto Nandi6, Ravi Salgia6 and Yi-Ching Wang1* Abstract Background: Cancer-related genes show racial differences. Therefore, identification and characterization of DNA copy number alteration regions in different racial groups helps to dissect the mechanism of tumorigenesis. Methods: Array-comparative genomic hybridization (array-CGH) was analyzed for DNA copy number profile in 40 Asian and 20 Caucasian lung cancer patients. Three methods including MetaCore analysis for disease and pathway correlations, concordance analysis between array-CGH database and the expression array database, and literature search for copy number variation genes were performed to select novel lung cancer candidate genes. Four candidate oncogenes were validated for DNA copy number and mRNA and protein expression by quantitative polymerase chain reaction (qPCR), chromogenic in situ hybridization (CISH), reverse transcriptase-qPCR (RT-qPCR), and immunohistochemistry (IHC) in more patients. Results: We identified 20 chromosomal imbalance regions harboring 459 genes for Caucasian and 17 regions containing 476 genes for Asian lung cancer patients. Seven common chromosomal imbalance regions harboring 117 genes, included gain on 3p13-14, 6p22.1, 9q21.13, 13q14.1, and 17p13.3; and loss on 3p22.2-22.3 and 13q13.3 were found both in Asian and Caucasian patients. -

Open Data for Differential Network Analysis in Glioma
International Journal of Molecular Sciences Article Open Data for Differential Network Analysis in Glioma , Claire Jean-Quartier * y , Fleur Jeanquartier y and Andreas Holzinger Holzinger Group HCI-KDD, Institute for Medical Informatics, Statistics and Documentation, Medical University Graz, Auenbruggerplatz 2/V, 8036 Graz, Austria; [email protected] (F.J.); [email protected] (A.H.) * Correspondence: [email protected] These authors contributed equally to this work. y Received: 27 October 2019; Accepted: 3 January 2020; Published: 15 January 2020 Abstract: The complexity of cancer diseases demands bioinformatic techniques and translational research based on big data and personalized medicine. Open data enables researchers to accelerate cancer studies, save resources and foster collaboration. Several tools and programming approaches are available for analyzing data, including annotation, clustering, comparison and extrapolation, merging, enrichment, functional association and statistics. We exploit openly available data via cancer gene expression analysis, we apply refinement as well as enrichment analysis via gene ontology and conclude with graph-based visualization of involved protein interaction networks as a basis for signaling. The different databases allowed for the construction of huge networks or specified ones consisting of high-confidence interactions only. Several genes associated to glioma were isolated via a network analysis from top hub nodes as well as from an outlier analysis. The latter approach highlights a mitogen-activated protein kinase next to a member of histondeacetylases and a protein phosphatase as genes uncommonly associated with glioma. Cluster analysis from top hub nodes lists several identified glioma-associated gene products to function within protein complexes, including epidermal growth factors as well as cell cycle proteins or RAS proto-oncogenes. -

Comprehensive Biological Information Analysis of PTEN Gene in Pan-Cancer
Comprehensive biological information analysis of PTEN gene in pan-cancer Hang Zhang Shanghai Medical University: Fudan University https://orcid.org/0000-0002-5853-7754 Wenhan Zhou Shanghai Medical University: Fudan University Xiaoyi Yang Shanghai Jiao Tong University School of Medicine Shuzhan Wen Shanghai Medical University: Fudan University Baicheng Zhao Shanghai Medical University: Fudan University Jiale Feng Shanghai Medical University: Fudan University Shuying Chen ( [email protected] ) https://orcid.org/0000-0002-9215-9777 Primary research Keywords: PTEN, correlated genes, TCGA, GEPIA, UALCAN, GTEx, expression, cancer Posted Date: April 12th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-388887/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/21 Abstract Background PTEN is a multifunctional tumor suppressor gene mutating at high frequency in a variety of cancers. However, its expression in pan-cancer, correlated genes, survival prognosis, and regulatory pathways are not completely described. Here, we aimed to conduct a comprehensive analysis from the above perspectives in order to provide reference for clinical application. Methods we studied the expression levels in cancers by using data from TCGA and GTEx database. Obtain expression box plot from UALCAN database. Perform mutation analysis on the cBioportal website. Obtain correlation genes on the GEPIA website. Construct protein network and perform KEGG and GO enrichment analysis on the STRING database. Perform prognostic analysis on the Kaplan-Meier Plotter website. We also performed transcription factor prediction on the PROMO database and performed RNA-RNA association and RNA-protein interaction on the RNAup Web server and RPISEq. -
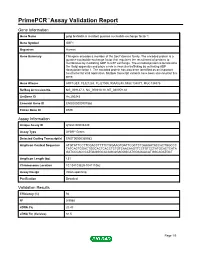
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name golgi brefeldin A resistant guanine nucleotide exchange factor 1 Gene Symbol GBF1 Organism Human Gene Summary This gene encodes a member of the Sec7 domain family. The encoded protein is a guanine nucleotide exchange factor that regulates the recruitment of proteins to membranes by mediating GDP to GTP exchange. The encoded protein is localized to the Golgi apparatus and plays a role in vesicular trafficking by activating ADP ribosylation factor 1. The encoded protein has also been identified as an important host factor for viral replication. Multiple transcript variants have been observed for this gene. Gene Aliases ARF1GEF, FLJ21263, FLJ21500, KIAA0248, MGC134877, MGC134878 RefSeq Accession No. NG_008147.1, NC_000010.10, NT_030059.13 UniGene ID Hs.290243 Ensembl Gene ID ENSG00000107862 Entrez Gene ID 8729 Assay Information Unique Assay ID qHsaCID0008440 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000369983 Amplicon Context Sequence ATGTATTCCTTCGACCTTTTCTGGAAGTGATTCGCTCTGAAGATACCACTGGCCC TATCACTGGACTGGCACTCACCTCTGTCAACAAGTTCCTGTCCTATGCACTCATA GATCCCACCCATGAGGGCACAGCAGAGGGCATGGAGAACATGGCAGATGCT Amplicon Length (bp) 131 Chromosome Location 10:104103829-104111062 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 94 R2 0.9988 cDNA Cq 20.43 cDNA Tm (Celsius) 84.5 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq 36.2 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay -

A Point Mutation in HIV-1 Integrase Redirects Proviral Integration Into
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.12.426369; this version posted January 12, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. A point mutation in HIV-1 integrase redirects proviral integration into centromeric repeats Shelby Winansa,b,c and Stephen P. Goffa.b,c# aDepartment of Biochemistry and Molecular Biophysics Columbia University Medical Center, New York, NY bDepartment of Microbiology and Immunology Columbia University Medical Center, New York, NY cHoward Hughes Medical Institute, Columbia University, New York, NY #Lead Contact: address correspondence to Stephen P. Goff, [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2021.01.12.426369; this version posted January 12, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Abstract 2 Retroviruses utilize the viral integrase (IN) protein to integrate a DNA copy of their 3 genome into the host chromosomal DNA. HIV-1 integration sites are highly biased towards 4 actively transcribed genes, likely mediated by binding of the IN protein to specific host 5 factors, particularly LEDGF, located at these gene regions. We here report a dramatic 6 redirection of integration site distribution induced by a single point mutation in HIV-1 IN. -

393LN V 393P 344SQ V 393P Probe Set Entrez Gene
393LN v 393P 344SQ v 393P Entrez fold fold probe set Gene Gene Symbol Gene cluster Gene Title p-value change p-value change chemokine (C-C motif) ligand 21b /// chemokine (C-C motif) ligand 21a /// chemokine (C-C motif) ligand 21c 1419426_s_at 18829 /// Ccl21b /// Ccl2 1 - up 393 LN only (leucine) 0.0047 9.199837 0.45212 6.847887 nuclear factor of activated T-cells, cytoplasmic, calcineurin- 1447085_s_at 18018 Nfatc1 1 - up 393 LN only dependent 1 0.009048 12.065 0.13718 4.81 RIKEN cDNA 1453647_at 78668 9530059J11Rik1 - up 393 LN only 9530059J11 gene 0.002208 5.482897 0.27642 3.45171 transient receptor potential cation channel, subfamily 1457164_at 277328 Trpa1 1 - up 393 LN only A, member 1 0.000111 9.180344 0.01771 3.048114 regulating synaptic membrane 1422809_at 116838 Rims2 1 - up 393 LN only exocytosis 2 0.001891 8.560424 0.13159 2.980501 glial cell line derived neurotrophic factor family receptor alpha 1433716_x_at 14586 Gfra2 1 - up 393 LN only 2 0.006868 30.88736 0.01066 2.811211 1446936_at --- --- 1 - up 393 LN only --- 0.007695 6.373955 0.11733 2.480287 zinc finger protein 1438742_at 320683 Zfp629 1 - up 393 LN only 629 0.002644 5.231855 0.38124 2.377016 phospholipase A2, 1426019_at 18786 Plaa 1 - up 393 LN only activating protein 0.008657 6.2364 0.12336 2.262117 1445314_at 14009 Etv1 1 - up 393 LN only ets variant gene 1 0.007224 3.643646 0.36434 2.01989 ciliary rootlet coiled- 1427338_at 230872 Crocc 1 - up 393 LN only coil, rootletin 0.002482 7.783242 0.49977 1.794171 expressed sequence 1436585_at 99463 BB182297 1 - up 393 -

Pharmacogenomics of Ventricular Conduction in Multi-Ethnic Populations Amanda Seyerle Dissertation Committee
Pharmacogenomics of Ventricular Conduction in Multi-Ethnic Populations Amanda Seyerle Dissertation Committee: Christy Avery (chair) Kari North Eric Whitsel Til Stürmer Craig Lee 1 TABLE OF CONTENTS Page LIST OF TABLES ...................................................................................................................................... v LIST OF FIGURES ..................................................................................................................................... vi LIST OF ABBREVIATIONS ..................................................................................................................... vii LIST OF GENE NAMES ............................................................................................................................ ix 1. Overview .............................................................................................................................................. 1 2. Specific Aims ....................................................................................................................................... 3 3. Background and Significance ............................................................................................................ 4 A. Ventricular Conduction ......................................................................................................................... 4 A.1. Electrical Conduction of the Heart ................................................................................................ 4 A.1.1. Sodium Channels.................................................................................................................. -

Gwas Meta-Analysis of Intelligence 1
bioRxiv preprint doi: https://doi.org/10.1101/184853; this version posted September 6, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. GWAS META-ANALYSIS OF INTELLIGENCE 1 GWAS meta-analysis (N=279,930) identifies new genes and functional links to intelligence Jeanne E Savage1#, Philip R Jansen1,2#, Sven Stringer1, Kyoko Watanabe1, Julien Bryois3, Christiaan A de Leeuw1, Mats Nagel1, Swapnil Awasthi4, Peter B Barr5, Jonathan R I Coleman6,7, Katrina L Grasby8, Anke R Hammerschlag1, Jakob Kaminski4,9, Robert Karlsson3, Eva Krapohl6, Max Lam10, Marianne Nygaard11,12, Chandra A Reynolds13, Joey W Trampush14, Hannah Young15, Delilah Zabaneh6, Sara Hägg3, Narelle K Hansell16, Ida K Karlsson3, Sten Linnarsson17, Grant W Montgomery8,18, Ana B Muñoz-Manchado17, Erin B Quinlan6, Gunter Schumann6, Nathan Skene17, Bradley T Webb19,20,21, Tonya White2, Dan E Arking22, Deborah K Attix23,24, Dimitrios Avramopoulos22,25, Robert M Bilder26, Panos Bitsios27, Katherine E Burdick28,29,30, Tyrone D Cannon31, Ornit Chiba-Falek32, Andrea Christoforou23, Elizabeth T Cirulli33, Eliza Congdon26, Aiden Corvin34, Gail Davies35, 36, Ian J Deary35,36, Pamela DeRosse37, Dwight Dickinson38, Srdjan Djurovic39,40, Gary Donohoe41, Emily Drabant Conley42, Johan G Eriksson43,44, Thomas Espeseth45,46, Nelson A Freimer26, Stella Giakoumaki47, Ina Giegling48, Michael Gill34, David