Proquest Dissertations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Te2, Part Iii
TERMINOLOGIA EMBRYOLOGICA Second Edition International Embryological Terminology FIPAT The Federative International Programme for Anatomical Terminology A programme of the International Federation of Associations of Anatomists (IFAA) TE2, PART III Contents Caput V: Organogenesis Chapter 5: Organogenesis (continued) Systema respiratorium Respiratory system Systema urinarium Urinary system Systemata genitalia Genital systems Coeloma Coelom Glandulae endocrinae Endocrine glands Systema cardiovasculare Cardiovascular system Systema lymphoideum Lymphoid system Bibliographic Reference Citation: FIPAT. Terminologia Embryologica. 2nd ed. FIPAT.library.dal.ca. Federative International Programme for Anatomical Terminology, February 2017 Published pending approval by the General Assembly at the next Congress of IFAA (2019) Creative Commons License: The publication of Terminologia Embryologica is under a Creative Commons Attribution-NoDerivatives 4.0 International (CC BY-ND 4.0) license The individual terms in this terminology are within the public domain. Statements about terms being part of this international standard terminology should use the above bibliographic reference to cite this terminology. The unaltered PDF files of this terminology may be freely copied and distributed by users. IFAA member societies are authorized to publish translations of this terminology. Authors of other works that might be considered derivative should write to the Chair of FIPAT for permission to publish a derivative work. Caput V: ORGANOGENESIS Chapter 5: ORGANOGENESIS -

Cardiovascular System Heart Development Cardiovascular System Heart Development
Cardiovascular System Heart Development Cardiovascular System Heart Development In human embryos, the heart begins to beat at approximately 22-23 days, with blood flow beginning in the 4th week. The heart is one of the earliest differentiating and functioning organs. • This emphasizes the critical nature of the heart in distributing blood through the vessels and the vital exchange of nutrients, oxygen, and wastes between the developing baby and the mother. • Therefore, the first system that completes its development in the embryo is called cardiovascular system. https://www.slideshare.net/DrSherifFahmy/intraembryonic-mesoderm-general-embryology Mesoderm is one of the three • Connective tissue primary germ layers that • Smooth and striated muscle • Cardiovascular System differentiates early in • Kidneys development that collectively • Spleen • Genital organs, ducts gives rise to all subsequent • Adrenal gland cortex tissues and organs. The cardiovascular system begins to develop in the third week of gestation. Blood islands develop in the newly formed mesoderm, and consist of (a) a central group of haemoblasts, the embryonic precursors of blood cells; (b) endothelial cells. Development of the heart and vascular system is often described together as the cardiovascular system. Development begins very early in mesoderm both within (embryonic) and outside (extra embryonic, vitelline, umblical and placental) the embryo. Vascular development occurs in many places. • Blood islands coalesce to form a vascular plexus. Preferential channels form arteries and veins. • Day 17 - Blood islands form first in the extra-embryonic mesoderm • Day 18 - Blood islands form next in the intra-embryonic mesoderm • Day 19 - Blood islands form in the cardiogenic mesoderm and coalesce to form a pair of endothelial heart tubes Development of a circulation • A circulation is established during the 4th week after the myocardium is differentiated. -

Truncus Arteriosus Communis: Report of Three Cases and Review of Literature
Truncus arteriosus communis: report of three cases and review of literature Henriette Poaty1,2, Fanny Pelluard3, Gwenaelle André3, Brigitte Maugey-Laulom4, Dominique Carles3 1. Histology-Embryology and Genetic Laboratory, Faculty of Health Sciences, BP 2672, Marien Ngouabi University, Brazzaville, Congo. 2. National Research Institute on Health Sciences, Brazzaville, Congo 3. Department of Fetopathology, CHU Pellegrin, place Amélie Raba, 33076 Bordeaux cedex France 4. Fetal Imaging Unit, Maternity, CHU Pellegrin, place Amélie Raba, 33076 Bordeaux, France Emails: Henriette Poaty- [email protected] Fanny Pelluard - [email protected] Gwenaelle André- [email protected] Brigitte Maugey-Laulom- [email protected] Dominique Carles- [email protected] Abstract Background: Truncus arteriosus communis (TAC) is a congenital heart defect in which the physiologic arterial common trunk was not divided into aorta and pulmonary artery trunk. Objectives: In this paper, we report on three observed cases from which we looked for (in conjunction with literature review) the different causes of TAC many of which have genetic origins. Methods: We collected three clinical files of fetuses having a TAC. Two of them were examinated after a medical termination of pregnancy motivated by severe cardiopathy. The malformation had been diagnosed based on different techniques: echocardi- ography, skeletal radiography, arteriography, fetal autopsy, karyotype and fluorescence in situ hybridization (FISH). Results: Imaging and fetopathological examination revealed the presence of TAC type 3 and 4 in the Van Praaghs classification. FISH analysis showed a 22q11.2 deletion in one fetus in favour of Digeorge syndrome. The karyotype analysis performed in two cases was normal. Conclusion: Truncus arteriosus is a rare pathology caused by numerous etiologies from which many of them have genetic ori- gin. -
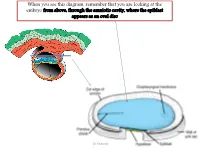
Embryo a the Development of the Heart
When you see this diagram, remember that you are looking at the embryo from above, through the amniotic cavity, where the epiblast appears as an oval disc Dr. Shatarat Dr. Shatarat Why the embryo needs the vascular system? When it appears? Where it appears? with later contributions from neural crest mesenchyme Dr. Shatarat The Cardiac progenitor cells migrate from the Epiblast Through In a Cranial direction on Primitive streak each side of the notochordal process and around the prechordal plate 4 Dr. Shatarat into the splanchnic layer of the lateral plate mesoderm Dr. Shatarat into the splanchnic layer of the lateral plate mesoderm Dr. Shatarat The cells from both sides meet cranially to form the Primary Heart Field (PHF) These cells will form : • The atria • Left ventricle • Part of right ventricle • The remainder of the right ventricle • outflow tract (conus cordis and truncus arteriosus) Are derived from the Secondary Heart Field (SHF) Dr. Shatarat ONE-SOMITE AND TWO-SOMITE STAGES Dr. Shatarat Paired endothelial strands ANGIOBLASTIC CORDS appear in the cardiogenic mesoderm during the third week of development Dr. Shatarat Dr. Shatarat These cords canalize to form two heart tubes that soon fuse as embryo folds laterally to form a single heart tube late in the third week Two hearts tubs The two tubs are Fused Single heart tube is formed As the embryo folds laterally Dr. Shatarat Dr. Shatarat In addition to the cardiogenic region, other blood islands appear bilaterally, parallel and close to the midline of the embryonic shield. These islands form a pair of longitudinal vessels, the dorsal aortae. -

Findings from Chick Embryo Studies
Journal of Cardiovascular Development and Disease Review Mechanosensitive Pathways in Heart Development: Findings from Chick Embryo Studies Maha Alser 1,2, Samar Shurbaji 1 and Huseyin C. Yalcin 1,* 1 Biomedical Research Center, Qatar University, Doha P.O. Box 2713, Qatar; [email protected] (M.A.); [email protected] (S.S.) 2 College of Health and Life Sciences, Hamad Bin Khalifa University, Doha P.O. Box 34110, Qatar * Correspondence: [email protected]; Tel.: +974-4403-7719 Abstract: The heart is the first organ that starts to function in a developing embryo. It continues to undergo dramatic morphological changes while pumping blood to the rest of the body. Genetic regulation of heart development is partly governed by hemodynamics. Chick embryo is a major animal model that has been used extensively in cardiogenesis research. To reveal mechanosensitive pathways, a variety of surgical interferences and chemical treatments can be applied to the chick embryo to manipulate the blood flow. Such manipulations alter expressions of mechanosensitive genes which may anticipate induction of morphological changes in the developing heart. This paper aims to present different approaches for generating clinically relevant disturbed hemodynamics conditions using this embryonic chick model and to summarize identified mechanosensitive genes using the model, providing insights into embryonic origins of congenital heart defects. Keywords: chick embryo; hemodynamics; mechanobiology; mechanosensitive pathways; surgical interferences; left atrial ligation; outflow tract banding; vitelline vein ligation; chemical treatment; gene expression Citation: Alser, M.; Shurbaji, S.; Yalcin, H.C. Mechanosensitive Pathways in Heart Development: Findings from Chick Embryo Studies. 1. Introduction J. Cardiovasc. Dev. -

6. Heart and Circulatory System I
6. HEART AND CIRCULATORY SYSTEM I Dr. Taube P. Rothman P&S 12-520 [email protected] 212-305-7930 RECOMMENDED READING: Larsen Human Embryology, 3rd Edition, pp. 195-199; 157-169 top left; 172-174; bottom 181-182; 187-top 189, Simbryo-cardiovascular system SUMMARY: The circulatory system, consisting of heart, blood vessels, and blood cells is the first functional organ to develop. This lecture will focus on the formation of the embryonic vasculature, the origin and formation of the early heart tube and primitive cardiac chambers, cardiac looping, and the primitive circulation. Between the 5th - 8th week of embryonic development, the tubular heart is remodeled into a four chambered structure. We will see how right and left atrioventricular canals connect each atrium with its respective ventricle, and how the atrial septum and definitive right and left atria form. We will also see why the great veins deliver blood to the right atrium while the pulmonary veins empty into the left. GLOSSARY: Angioblasts: precursors of blood vessels Angiogenesis: lengthening, branching, sprouting and remodeling of embryonic blood vessels Aortic arches: paired arteries surrounding the pharynx; portions will contribute to formation of the great arterial vessels Blood islands: clusters of cells in the yolk sac, connecting stalk and chorionic villi that form primitive blood vessels Cardiac jelly: gelatinous extracellular matrix that forms the middle layer of the heart tube Ductus venosus: shunts most of the blood in the umbilical vein into the inferior vena cava -

Development of CVS I
Development of CVS I Systemic Embryology Heart development (cardiogenesis) • Prenatal development of the human heart • Begins with the formation of two endocardial tubes • These tubes merge to form the tubular heart, also called the primitive heart tube • Primitive heart tube loops and septates into four chambers and paired arterial trunks that form the adult heart. 2 3 Early development of the heart 4 • First major system to function in the embryo. • Primordial heart and vascular system appear in the middle of the third week (Day 21or 22). - The rapidly growing embryo can no longer satisfy its nutritional and oxygen requirements by diffusion alone. - A need for an efficient method of acquiring oxygen and nutrients from the maternal blood and disposing of carbon dioxide and waste products. 5 Derived from: • Splanchnic mesoderm, which forms the primordium of the heart • Paraxial and lateral mesoderm • Pharyngeal mesoderm • Neural crest cells 6 • In the splanchnopleuric mesenchyme on either side of the neural plate, a horseshoe-shaped area develops as the cardiogenic region • Formed from cardiac myoblasts and blood islands • By day 19, an endocardial tube begins to develop in each side of this region • Tubes grow, converge and merge to form a single tube – the tubular heart by programmed cell death 7 • Cardiac progenitor cells in the epiblast, lateral to the primitive streak migrate through the streak. • Cardiogenic region develops cranially and laterally to the neural plate • As embryonic folding continues, the two endocardial tubes are pushed into the thoracic cavity, where they fuse together at about the 22nd day 8 • Heart develop near the head of the embryo in the cardiogenic region • Cells in the splanchnic layer of the lateral plate mesoderm are induced by pharyngeal endoderm to form cardiac myoblasts. -

Cardiovascular System - Accessscience from Mcgraw-Hill Education
Cardiovascular system - AccessScience from McGraw-Hill Education http://accessscience.com/content/109900 (http://accessscience.com/) Article by: Weichert, Charles K. College of Arts and Sciences, University of Cincinnati, Cincinnati, Ohio. Copenhaver, W. M. College of Physicians and Surgeons, Columbia University, New York; Department of Biological Structures, School of Medicine, University of Miami, Miami, Florida. Ebert, James D. Department of Embryology, Carnegie Institution, Washington, DC. Patten, Bradley M. Department of Anatomy, University of Michigan, Ann Arbor, Michigan. Jones, David R. Department of Zoology, University of British Columbia, Vancouver, Canada. Publication year: 2014 DOI: http://dx.doi.org/10.1036/1097-8542.109900 (http://dx.doi.org/10.1036/1097-8542.109900) Content Comparative Anatomy Embryogenesis of blood vessels Balancing ventricular output Heart Angiogenesis Human Postnatal Circulation Arterial system Circulatory system morphogenesis Pulmonary circuit and ductus Venous system Primitive venous system Physiological aspects of transition Comparative Embryology Functional Development of Heart Comparative Physiology Heart Contractions of the heart General physiology of circulation Tubular heart formation Heart-forming areas Microcirculation Cardiac loop and regional development Contractile proteins Heart Formation of definitive heart Synthesis of contractile proteins Arteries Partitioning of mammalian heart Action of inhibitors Venous system Division of atrium and ventricles Human Fetal Circulation at Term Bibliography -

The Role of Endoglin in Endothelial and Mesenchymal Cells During Development, Maintenance and Repair of the Heart
The Role of Endoglin in Endothelial and Mesenchymal cells during Development, Maintenance and Repair of the Heart Esha Singh Thesis submitted for the degree of Doctor of Philosophy Institute of Genetic Medicine Faculty of Medical Sciences Newcastle University December 2019 Abstract Mutations in the endoglin gene lead to Haemorrhagic Telangiectasia Type 1 (HHT1), an inherited vascular disease, demonstrating the importance of endoglin in the vasculature. Haplo- deficiency of endoglin also leads to reduced angiogenesis in mice in several pathologies, indicating a pro-angiogenic role for endoglin. However, the precise role of endoglin in development and maintenance of the vasculature is still unclear. The aim of this project therefore was to investigate the role of endoglin during vascular development and maintenance and whether its pro-angiogenic properties can be used to enhance cardiac recovery following myocardial infarction (MI). Using immunofluorescence staining of mouse cardiac tissue, I showed high endoglin expression in coronary veins endothelial cells (ECs), capillaries & endocardium and weak expression in coronary arteries ECs, valve mesenchyme & in epicardially derived cells (EPDCs) during development. I hypothesised that during cardiac development endoglin is essential for EC proliferation and migration, and plays a key role during EPDC proliferation, migration and differentiation. Induced knockdown of endoglin in murine epicardial cells using Wt1-Cre led to a reduced number of EPDCs in E15.5 hearts but did not lead to any detectable vascular defects in E17.5 hearts. In contrast, EC-specific endoglin knockdown in the first week of postnatal life (which is known to lead to retinal AVMs) was associated with the development of eccentric cardiomyopathy and cardiomyocytes (CM) hypertrophy, but no obvious coronary vasculature defects. -

Right Ventricular Phenotype, Function, and Failure: a Journey from Evolution to Clinics
Heart Failure Reviews https://doi.org/10.1007/s10741-020-09982-4 Right ventricular phenotype, function, and failure: a journey from evolution to clinics Yannick J. H. J. Taverne1,2,3 & Amir Sadeghi1 & Beatrijs Bartelds4 & Ad J. J. C. Bogers1 & Daphne Merkus2 # The Author(s) 2020 Abstract The right ventricle has long been perceived as the “low pressure bystander” of the left ventricle. Although the structure consists of, at first glance, the same cardiomyocytes as the left ventricle, it is in fact derived from a different set of precursor cells and has a complex three-dimensional anatomy and a very distinct contraction pattern. Mechanisms of right ventricular failure, its detection and follow-up, and more specific different responses to pressure versus volume overload are still incompletely understood. In order to fully comprehend right ventricular form and function, evolutionary biological entities that have led to the specifics of right ventricular physiology and morphology need to be addressed. Processes responsible for cardiac formation are based on very ancient cardiac lineages and within the first few weeks of fetal life, the human heart seems to repeat cardiac evolution. Furthermore, it appears that most cardiogenic signal pathways (if not all) act in combination with tissue-specific transcriptional cofactors to exert inductive responses reflecting an important expansion of ancestral regulatory genes throughout evolution and eventually cardiac complexity. Such molecular entities result in specific biomechanics of the RV that differs from that of the left ventricle. It is clear that sole descriptions of right ventricular contraction patterns (and LV contraction patterns for that matter) are futile and need to be addressed into a bigger multilayer three-dimensional picture. -

447 Mmps in Cardiovascular Development and Disease
[Frontiers in Bioscience 11, 447-478, January 1, 2006] MMPs - Role in Cardiovascular Development and Disease Philip R. Brauer Department of Biomedical Sciences, Creighton University School of Medicine, Omaha, Nebraska 68178 TABLE OF CONTENTS 1. Abstract 2. Introduction 3. MMPs and MMP Inhibitors 4. MMP Activation 5. Overview of MMP Functions 5.1. Cell-Extracellular Matrix Adhesion, Migration, and Invasion 5.2. MMPs and Epithelial-to-Mesenchymal Transitions 5.3. MMPs and Cell Signaling 5.4. MMP Deficient Animals 6. MMPs and TIMPs in Cardiovascular Morphogenesis 6.1. Embryonic Vasculogenesis and Angiogenesis 6.2. MMPs and TIMPs in Heart Morphogenesis 6.2.1. MMPs and Cardiac Tube Formation and Looping 6.2.1. Heart Septation 6.2.3. Embryonic and Fetal Cardiac Remodeling 7. MMPs and TIMPs in Cardiovascular Disease 7.1. MMPs and Atherosclerosis 7.1.1. Plaque Development, Intimal Thickening, and Plaque Stability 7.1.2. Restenosis 7.2. MMPs and Aneurysms 7.3. MMPs and Progressive Heart Failure 8. Perspectives 9. Acknowledgements 10. References 1. ABSTRACT 2. INTRODUCTION Matrix metalloproteinases (MMPs) are a family Matrix metalloproteinases are proteolytic of proteolytic enzymes important in the degradation and enzymes whose function is primarily viewed as being the turnover of extracellular matrix (ECM) components. MMPs degradation and turnover of ECM components. However, and their inhibitors play major roles not only in ECM MMPs and their inhibitors also play key roles in regulating degradation but also in mediating cell-cell adhesion, cell many fundamental cell processes including regulation of migration and invasion, cell proliferation and apoptosis, cell growth, cell adhesion, cell migration and invasion, cell tissue remodeling, and growth factor and cytokine death, and tissue remodeling events. -

Anas Platyrhynchos, and Ducks Offer Advantages Compared to Other Model Organisms, Such As Their Larger Size and Durability
Effects of alcohol on the development of the cardiovascular system in Pekin Ducks (Anas platyrhynchos): An assessment of current empirical findings and the development of a research protocol utilizing Pekin Ducks A project completed in partial fulfillment of the requirements for the Honors Program By Josephine McKean May 8, 2021 Department of Biological and Environmental Sciences Capital University Approved by Nancy J Swails Name, Advisor __________John Mersfelder______________________________________ Name, Department Chair Accepted by ________________________________________________ Name, Director, Capital University Honors Program 1 Copyrighted by Josephine McKean 2021 2 Abstract Fetal alcohol syndrome is a serious condition that affects the development of fetuses with irreversible effects that can impact individuals throughout their lives. The cardiovascular system is one example of an organ system in which abnormalities caused by alcohol can occur. The heart is one of the first structures to be formed, and heart development is highly conserved among amniotes. There are difficulties studying the effects of ethanol on human embryos due to ethical concerns; as a result, the use of animal models, particularly avian models, is widely used. The effects of ethanol have not been widely studied on Pekin ducks, Anas platyrhynchos, and ducks offer advantages compared to other model organisms, such as their larger size and durability. The purpose of this study was to develop a method for testing the effects of ethanol on the development of the heart and cardiovascular system in ducks. The development of the cardiovascular system occurs over several stages of development, and treatment of ethanol at different stages leads to various potential abnormalities of heart structure and function.