Right Ventricular Phenotype, Function, and Failure: a Journey from Evolution to Clinics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Te2, Part Iii
TERMINOLOGIA EMBRYOLOGICA Second Edition International Embryological Terminology FIPAT The Federative International Programme for Anatomical Terminology A programme of the International Federation of Associations of Anatomists (IFAA) TE2, PART III Contents Caput V: Organogenesis Chapter 5: Organogenesis (continued) Systema respiratorium Respiratory system Systema urinarium Urinary system Systemata genitalia Genital systems Coeloma Coelom Glandulae endocrinae Endocrine glands Systema cardiovasculare Cardiovascular system Systema lymphoideum Lymphoid system Bibliographic Reference Citation: FIPAT. Terminologia Embryologica. 2nd ed. FIPAT.library.dal.ca. Federative International Programme for Anatomical Terminology, February 2017 Published pending approval by the General Assembly at the next Congress of IFAA (2019) Creative Commons License: The publication of Terminologia Embryologica is under a Creative Commons Attribution-NoDerivatives 4.0 International (CC BY-ND 4.0) license The individual terms in this terminology are within the public domain. Statements about terms being part of this international standard terminology should use the above bibliographic reference to cite this terminology. The unaltered PDF files of this terminology may be freely copied and distributed by users. IFAA member societies are authorized to publish translations of this terminology. Authors of other works that might be considered derivative should write to the Chair of FIPAT for permission to publish a derivative work. Caput V: ORGANOGENESIS Chapter 5: ORGANOGENESIS -

Fetal Blood Flow and Genetic Mutations in Conotruncal Congenital Heart Disease
Journal of Cardiovascular Development and Disease Review Fetal Blood Flow and Genetic Mutations in Conotruncal Congenital Heart Disease Laura A. Dyer 1 and Sandra Rugonyi 2,* 1 Department of Biology, University of Portland, Portland, OR 97203, USA; [email protected] 2 Department of Biomedical Engineering, Oregon Health & Science University, Portland, OR 97239, USA * Correspondence: [email protected] Abstract: In congenital heart disease, the presence of structural defects affects blood flow in the heart and circulation. However, because the fetal circulation bypasses the lungs, fetuses with cyanotic heart defects can survive in utero but need prompt intervention to survive after birth. Tetralogy of Fallot and persistent truncus arteriosus are two of the most significant conotruncal heart defects. In both defects, blood access to the lungs is restricted or non-existent, and babies with these critical conditions need intervention right after birth. While there are known genetic mutations that lead to these critical heart defects, early perturbations in blood flow can independently lead to critical heart defects. In this paper, we start by comparing the fetal circulation with the neonatal and adult circulation, and reviewing how altered fetal blood flow can be used as a diagnostic tool to plan interventions. We then look at known factors that lead to tetralogy of Fallot and persistent truncus arteriosus: namely early perturbations in blood flow and mutations within VEGF-related pathways. The interplay between physical and genetic factors means that any one alteration can cause significant disruptions during development and underscore our need to better understand the effects of both blood flow and flow-responsive genes. -

Of the Bulbus Cordis
Dr.Amjad Sahatarat 1 Two opposing ridges are developed in the walls of the Truncus Arteriosus Called Truncal ridges And in the walls of Bulbus Cordis Called Bulbar ridges The bulbus cordis is also some times named conus and therefore The ridges developed inside it are also called conal. And with those developed in the truncus arteriosus they also together called These ridges are derived mainly from the Conotruncal Ridges neural crest When these ridges are fused with each other, They form Septa So ridges developed in the truncus arteriosus ridges developed in the lumen of the bulbus after their fusion are called cordis after their fusion are called Truncal septum bulbar septum We will study first of all the bulbar septum The Distal bulbar septum The Proximal bulbar septum The proximal bulbar septum shares A) in closing the interventricular foramen The proximal bulbar septum also B) incorporated into the walls of the definitive ventricles in several ways: into the infundibulum and the vestibule In the right ventricle, the bulbus cordis is represented by the conus arteriosus (infundibulum), which gives origin to the pulmonary trunk Dr.Amjad Sahatarat 6 In the left ventricle, the bulbus cordis forms the walls of the aortic vestibule the part of the ventricular cavity just inferior to the aortic valve. The distal bulbar septum 1- Four endocardinal cushions ( one anterior, one posterior, and two lateral right and left) are developed in the distal part of the bulbus cordis. 2- A ridge is developed in the middle of each of the two lateral cushions. It should be noted that the development of these ridges will divide each of the lateral cushions into two Dr.Amjad Sahatarat 9 3-These ridges will fuse to form a complete septum called the distal bulbar septum. -

Genetic and Flow Anomalies in Congenital Heart Disease
Published online: 2021-05-10 AIMS Genetics, 3(3): 157-166. DOI: 10.3934/genet.2016.3.157 Received: 01 July 2016 Accepted: 16 August 2016 Published: 23 August 2016 http://www.aimspress.com/journal/Genetics Review Genetic and flow anomalies in congenital heart disease Sandra Rugonyi* Department of Biomedical Engineering, Oregon Health & Science University, 3303 SW Bond Ave. M/C CH13B, Portland, OR 97239, USA * Correspondence: Email: [email protected]; Tel: +1-503-418-9310; Fax: +1-503-418-9311. Abstract: Congenital heart defects are the most common malformations in humans, affecting approximately 1% of newborn babies. While genetic causes of congenital heart disease have been studied, only less than 20% of human cases are clearly linked to genetic anomalies. The cause for the majority of the cases remains unknown. Heart formation is a finely orchestrated developmental process and slight disruptions of it can lead to severe malformations. Dysregulation of developmental processes leading to heart malformations are caused by genetic anomalies but also environmental factors including blood flow. Intra-cardiac blood flow dynamics plays a significant role regulating heart development and perturbations of blood flow lead to congenital heart defects in animal models. Defects that result from hemodynamic alterations recapitulate those observed in human babies, even those due to genetic anomalies and toxic teratogen exposure. Because important cardiac developmental events, such as valve formation and septation, occur under blood flow conditions while the heart is pumping, blood flow regulation of cardiac formation might be a critical factor determining cardiac phenotype. The contribution of flow to cardiac phenotype, however, is frequently ignored. -

Prenatal Development Timeline
Prenatal Development Timeline Nervous Cardiovascular Muscular Early Events Special Senses Respiratory Skeletal Growth Parameters Blood & Immune Gastrointestinal Endocrine General Skin/Integument Renal/Urinary Reproductive Movement Unit 1: The First Week Day 0 — — Embryonic period begins Fertilization resulting in zygote formation Day 1 — — Embryo is spherically shaped with 12 to 16 cells Day 1 - Day 1 — — Fertilization - development begins with a single-cell embryo!!! Day 2 — — Early pregnancy factor (EPF) Activation of the genome Zygote divides into two blastomeres (24 – 30 hours from start of fertilization) Day 4 — — Embryonic disc Free floating blastocyst Hypoblast & epiblast Inner cell mass See where the back and chest will be Day 5 — — Hatching blastocyst Day 6 — — Embryo attaches to wall of uterus 1 week — — Chorion Placenta begins to form Unit 2: 1 to 2 Weeks 1 week, 1 day — — Amnioblasts present; amnion and amniotic cavity formation begins Positive pregnancy test 1 week, 2 days — — Cells in womb engorged with nutrients 1 week, 4 days — — Longitudinal axis 1 week, 5 days — — Implantation complete Yolk sac 1 week, 6 days — — Primordial blood vessels Amnion with single cell layer Chorionic villi 2 weeks — — Yolk sac Yolk sac Unit 3: 2 to 3 Weeks 2 weeks, 1 day — — 3 germ layers Rostral-caudal orientation 2 weeks, 2 days — — Erythroblasts in yolk sac Three types of blood-forming cells in yolk sac Amnion with two cell layers Secondary villi www.ehd.org 1 of 10 2 weeks, 4 days — — Foregut, midgut, and hindgut Brain is first organ to -

Tetralogy of Fallot with Pulmonary Obstruction at the Level of the Conus Inlet a CASE REPORT
6 April 1974 S.-A. MEDIESE TYDSKRIF 677 Tetralogy of Fallot with Pulmonary Obstruction at the Level of the Conus Inlet A CASE REPORT T. MULLER SUMMARY in diameter could be seen. The right ventricle was enlarged and the thickness of the wall was 13,5 mm, compared with A case of Fallot's tetralogy is described in a Black male the 12,5 mm thickness of the left ventricle. The right who died of acute cardiac failure at the age of 17 years. ventricle communicated with the left ventricle through a The conus arteriosus was practically a separate chamber very large defect of the interventricular septum, which communicating with the right ventricle through a very easily admitted 3 fingers and which was straddled by the small ostium. The embryology of the truncus arteriosus aorta. The right ventricle was completely demarcated from the bulbus cordis is discussed in the light of the anomalies the infundibulum or conus arteriosus, the only connection described here. The question of maintenance of the pul being an ostium of 7,5 mm in diameter. monary circulation in the absence of an open ductus The conus arteriosus was a well-developed entity, both arteriosus is discussed. externally and internally (Figs 1 and 2). The interior of the conus arteriosus adjoining the right ventricle showed trabeculae carneae, but the upper portion leading to the S. Air. Med. J.• 48, 677 (1974). pulmonary valve was smooth. The pulmonary artery was reduced in size to half of that of the aort'i, and had only 2 valves (Fig. 2). -

EXTRACORONARY CARDIAC VEINS in the RAT1 the Present Work
EXTRACORONARY CARDIAC VEINS IN THE RAT1 MYRON H. HALPERN Department of Anatomy, Unit-ersity of Michigan, Ann Arbor SIX FIGURES The present work had its inception in the discovery of vessels around the rat’s heart which did not correspond to anything previously described in other mammals. These ves- sels are a system of veins which begin on the heart and terminate in the anterior venae cavae. Two major veins eom- prise this system, each of which crosses the midline to empty into the contralateral anterior vena cava. They drain the conal region of the right ventricle and the ventrocephalic region of the left ventricle. The term “extracoronary” cardiac veins has been applied to these vessels by the author because they originate on the heart and terminate in remote vessels not otherwise associated with the coronary circulation. Al- though this system has been found to exist in certain fishes and amphibians, to the author’s knowledge it has never been recognized in mammals. These findings seemed to warrant a more detailed study of the adult cardiac venous drainage of the rat. To supplement this portion of the investigation, an embryologic study was undertaken. Both the adult and the embryonic patterns of the cardiac drainage were com- pared with the patterns found in the above vertebrates and were interpreted on the basis of these comparisons. MATERIAL AND METHODS For this study, the venous system of 85 adult rats were injected with latex preparatory to dissection. Of this number, Portion of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the University of Michigan. -

Cardiogenesis with a Focus on Vasculogenesis and Angiogenesis
Received: 27 August 2019 | Revised: 4 February 2020 | Accepted: 20 February 2020 DOI: 10.1111/ahe.12549 SPECIAL ISSUE Cardiogenesis with a focus on vasculogenesis and angiogenesis Katrin Borasch1 | Kenneth Richardson2 | Johanna Plendl1 1Department of Veterinary Medicine, Institute of Veterinary Anatomy, Freie Abstract University Berlin, Berlin, Germany The initial intraembryonic vasculogenesis occurs in the cardiogenic mesoderm. Here, 2 College of Veterinary Medicine, School a cell population of proendocardial cells detaches from the mesoderm that subse- of Veterinary and Life Sciences, Murdoch University, Murdoch, WA, Australia quently generates the single endocardial tube by forming vascular plexuses. In the course of embryogenesis, the endocardium retains vasculogenic, angiogenic and Correspondence Johanna Plendl, Department of Veterinary haematopoietic potential. The coronary blood vessels that sustain the rapidly ex- Medicine, Institute of Veterinary Anatomy, panding myocardium develop in the course of the formation of the cardiac loop by Freie University Berlin, Berlin, Germany. Email: [email protected] vasculogenesis and angiogenesis from progenitor cells of the proepicardial serosa at the venous pole of the heart as well as from the endocardium and endothelial cells of Funding information Freie Universität Berlin the sinus venosus. Prospective coronary endothelial cells and progenitor cells of the coronary blood vessel walls (smooth muscle cells, perivascular cells) originate from different cell populations that are in close spatial as well as regulatory connection with each other. Vasculo- and angiogenesis of the coronary blood vessels are for a large part regulated by the epicardium and epicardium-derived cells. Vasculogenic and angiogenic signalling pathways include the vascular endothelial growth factors, the angiopoietins and the fibroblast growth factors and their receptors. -

Cardiovascular System Heart Development Cardiovascular System Heart Development
Cardiovascular System Heart Development Cardiovascular System Heart Development In human embryos, the heart begins to beat at approximately 22-23 days, with blood flow beginning in the 4th week. The heart is one of the earliest differentiating and functioning organs. • This emphasizes the critical nature of the heart in distributing blood through the vessels and the vital exchange of nutrients, oxygen, and wastes between the developing baby and the mother. • Therefore, the first system that completes its development in the embryo is called cardiovascular system. https://www.slideshare.net/DrSherifFahmy/intraembryonic-mesoderm-general-embryology Mesoderm is one of the three • Connective tissue primary germ layers that • Smooth and striated muscle • Cardiovascular System differentiates early in • Kidneys development that collectively • Spleen • Genital organs, ducts gives rise to all subsequent • Adrenal gland cortex tissues and organs. The cardiovascular system begins to develop in the third week of gestation. Blood islands develop in the newly formed mesoderm, and consist of (a) a central group of haemoblasts, the embryonic precursors of blood cells; (b) endothelial cells. Development of the heart and vascular system is often described together as the cardiovascular system. Development begins very early in mesoderm both within (embryonic) and outside (extra embryonic, vitelline, umblical and placental) the embryo. Vascular development occurs in many places. • Blood islands coalesce to form a vascular plexus. Preferential channels form arteries and veins. • Day 17 - Blood islands form first in the extra-embryonic mesoderm • Day 18 - Blood islands form next in the intra-embryonic mesoderm • Day 19 - Blood islands form in the cardiogenic mesoderm and coalesce to form a pair of endothelial heart tubes Development of a circulation • A circulation is established during the 4th week after the myocardium is differentiated. -
Development of Right Ventricle
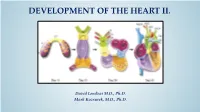
DEVELOPMENT OF THE HEART II. David Lendvai M.D., Ph.D. Mark Kozsurek, M.D., Ph.D. • Septation of the common atrioventricular (AV) orifice. • Formation of the interatrial septum. • Formation of the muscular interventricular septum. • Appearance of the membranous interventricular septum and the spiral aorticopulmonary septum. right left septum primum septum primum septum primum septum primum septum primum septum primum foramen primum foramen primum septum primum septum primum foramen primum foramen primum septum primum septum primum foramen secundum foramen secundum foramen primum foramen primum septum primum foramen secundum septum primum foramen secundum foramen primum foramen primum septum primum septum primum foramen secundum foramen secundum septum secundum septum secundum foramen secundum foramen ovale foramen ovale septum primum septum primum septum secundum septum secundum foramen secundum foramen ovale foramen ovale septum primum septum primum septum secundum septum secundum foramen secundum septum primum foramen ovale foramen ovale septum primum SUMMARY • The septation of the common atrium starts with the appearance of the crescent-shaped septum primum. The opening of this septum, the foramen primum, becomes progressively smaller. • Before the foramen primum completly closes, postero-superiorly several small openings appear on the septum primum. These perforations coalesce later and form the foramen secundum. • On the right side of the septum primum a new septum, the septum secundum, starts to grow. The orifice of the septum secundum is the foramen ovale. • Finally two crescent-like, incomplete, partially overlapping septa exist with one hole on each. Septum secundum is more rigid and the septum primum on its left side acts as a valve letting the blood flow exclusively from the right to the left. -

Truncus Arteriosus Communis: Report of Three Cases and Review of Literature
Truncus arteriosus communis: report of three cases and review of literature Henriette Poaty1,2, Fanny Pelluard3, Gwenaelle André3, Brigitte Maugey-Laulom4, Dominique Carles3 1. Histology-Embryology and Genetic Laboratory, Faculty of Health Sciences, BP 2672, Marien Ngouabi University, Brazzaville, Congo. 2. National Research Institute on Health Sciences, Brazzaville, Congo 3. Department of Fetopathology, CHU Pellegrin, place Amélie Raba, 33076 Bordeaux cedex France 4. Fetal Imaging Unit, Maternity, CHU Pellegrin, place Amélie Raba, 33076 Bordeaux, France Emails: Henriette Poaty- [email protected] Fanny Pelluard - [email protected] Gwenaelle André- [email protected] Brigitte Maugey-Laulom- [email protected] Dominique Carles- [email protected] Abstract Background: Truncus arteriosus communis (TAC) is a congenital heart defect in which the physiologic arterial common trunk was not divided into aorta and pulmonary artery trunk. Objectives: In this paper, we report on three observed cases from which we looked for (in conjunction with literature review) the different causes of TAC many of which have genetic origins. Methods: We collected three clinical files of fetuses having a TAC. Two of them were examinated after a medical termination of pregnancy motivated by severe cardiopathy. The malformation had been diagnosed based on different techniques: echocardi- ography, skeletal radiography, arteriography, fetal autopsy, karyotype and fluorescence in situ hybridization (FISH). Results: Imaging and fetopathological examination revealed the presence of TAC type 3 and 4 in the Van Praaghs classification. FISH analysis showed a 22q11.2 deletion in one fetus in favour of Digeorge syndrome. The karyotype analysis performed in two cases was normal. Conclusion: Truncus arteriosus is a rare pathology caused by numerous etiologies from which many of them have genetic ori- gin. -

Cardiovascular System Note: the Cardiovascular System Develops Early (Week 3), Enabling the Embryo to Grow Beyond the Short
Lymphatics: Lymph vessel formation is similar to blood angiogenesis. Lymphatics begin as lymph sacs in three regions: jugular (near brachiocephalic veins); cranial abdominal (future cysterna chyla); and iliac region. Lym- phatic vessels (ducts) form as outgrowths of the sacs. mesenchyme Lymph nodes are produced by localized mesoder- sinusoid lymph duct lumen mal invaginations that partition the vessel lumen into sinu- soids. The mesoderm develops a reticular framework within which mesodermal lymphocytes accumulate. The spleen and hemal nodes (in ruminants) invagination develop similar to the way lymph nodes develop. Lymph Node Formation Prior to birth, fetal circulation is designed for an in utero aqueous environment where the pla- centa oxygenates fetal blood. Suddenly, at birth... Three In-Utero Adjustments ductus Stretching and constriction of arteriosus umbilical arteries shifts fetal blood flow aortic arch from the placenta to the fetus. Reduced pulmonary trunk L atrium venous return through the (left) umbili- foramen ovale R cal vein and ductus venosus allows the atrium latter to gradually close (over a period caudal vena cava of days). Bradykinin released by expand- ductus venosus ing lungs and increased oxygen concen- tration in blood triggers constriction of aorta the ductus arteriosus which, over two liver months, is gradually converted to a fibrous structure, the ligamentum arte- umbilical v. riosum. portal v. The increased blood flow to the lungs and then to the left atrium equalizes pres- sure in the two atria, resulting in closure umbilical aa. of the foramen ovale that eventually grows permanent. 29 The cardiogenic area, the place where the embryonic heart originates, is located .