Soxf Factors Induce Notch1 Expression Via Direct Transcriptional Regulation During Early Arterial Development
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Of the Bulbus Cordis
Dr.Amjad Sahatarat 1 Two opposing ridges are developed in the walls of the Truncus Arteriosus Called Truncal ridges And in the walls of Bulbus Cordis Called Bulbar ridges The bulbus cordis is also some times named conus and therefore The ridges developed inside it are also called conal. And with those developed in the truncus arteriosus they also together called These ridges are derived mainly from the Conotruncal Ridges neural crest When these ridges are fused with each other, They form Septa So ridges developed in the truncus arteriosus ridges developed in the lumen of the bulbus after their fusion are called cordis after their fusion are called Truncal septum bulbar septum We will study first of all the bulbar septum The Distal bulbar septum The Proximal bulbar septum The proximal bulbar septum shares A) in closing the interventricular foramen The proximal bulbar septum also B) incorporated into the walls of the definitive ventricles in several ways: into the infundibulum and the vestibule In the right ventricle, the bulbus cordis is represented by the conus arteriosus (infundibulum), which gives origin to the pulmonary trunk Dr.Amjad Sahatarat 6 In the left ventricle, the bulbus cordis forms the walls of the aortic vestibule the part of the ventricular cavity just inferior to the aortic valve. The distal bulbar septum 1- Four endocardinal cushions ( one anterior, one posterior, and two lateral right and left) are developed in the distal part of the bulbus cordis. 2- A ridge is developed in the middle of each of the two lateral cushions. It should be noted that the development of these ridges will divide each of the lateral cushions into two Dr.Amjad Sahatarat 9 3-These ridges will fuse to form a complete septum called the distal bulbar septum. -

Tetralogy of Fallot with Pulmonary Obstruction at the Level of the Conus Inlet a CASE REPORT
6 April 1974 S.-A. MEDIESE TYDSKRIF 677 Tetralogy of Fallot with Pulmonary Obstruction at the Level of the Conus Inlet A CASE REPORT T. MULLER SUMMARY in diameter could be seen. The right ventricle was enlarged and the thickness of the wall was 13,5 mm, compared with A case of Fallot's tetralogy is described in a Black male the 12,5 mm thickness of the left ventricle. The right who died of acute cardiac failure at the age of 17 years. ventricle communicated with the left ventricle through a The conus arteriosus was practically a separate chamber very large defect of the interventricular septum, which communicating with the right ventricle through a very easily admitted 3 fingers and which was straddled by the small ostium. The embryology of the truncus arteriosus aorta. The right ventricle was completely demarcated from the bulbus cordis is discussed in the light of the anomalies the infundibulum or conus arteriosus, the only connection described here. The question of maintenance of the pul being an ostium of 7,5 mm in diameter. monary circulation in the absence of an open ductus The conus arteriosus was a well-developed entity, both arteriosus is discussed. externally and internally (Figs 1 and 2). The interior of the conus arteriosus adjoining the right ventricle showed trabeculae carneae, but the upper portion leading to the S. Air. Med. J.• 48, 677 (1974). pulmonary valve was smooth. The pulmonary artery was reduced in size to half of that of the aort'i, and had only 2 valves (Fig. 2). -

EXTRACORONARY CARDIAC VEINS in the RAT1 the Present Work
EXTRACORONARY CARDIAC VEINS IN THE RAT1 MYRON H. HALPERN Department of Anatomy, Unit-ersity of Michigan, Ann Arbor SIX FIGURES The present work had its inception in the discovery of vessels around the rat’s heart which did not correspond to anything previously described in other mammals. These ves- sels are a system of veins which begin on the heart and terminate in the anterior venae cavae. Two major veins eom- prise this system, each of which crosses the midline to empty into the contralateral anterior vena cava. They drain the conal region of the right ventricle and the ventrocephalic region of the left ventricle. The term “extracoronary” cardiac veins has been applied to these vessels by the author because they originate on the heart and terminate in remote vessels not otherwise associated with the coronary circulation. Al- though this system has been found to exist in certain fishes and amphibians, to the author’s knowledge it has never been recognized in mammals. These findings seemed to warrant a more detailed study of the adult cardiac venous drainage of the rat. To supplement this portion of the investigation, an embryologic study was undertaken. Both the adult and the embryonic patterns of the cardiac drainage were com- pared with the patterns found in the above vertebrates and were interpreted on the basis of these comparisons. MATERIAL AND METHODS For this study, the venous system of 85 adult rats were injected with latex preparatory to dissection. Of this number, Portion of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the University of Michigan. -

Cardiogenesis with a Focus on Vasculogenesis and Angiogenesis
Received: 27 August 2019 | Revised: 4 February 2020 | Accepted: 20 February 2020 DOI: 10.1111/ahe.12549 SPECIAL ISSUE Cardiogenesis with a focus on vasculogenesis and angiogenesis Katrin Borasch1 | Kenneth Richardson2 | Johanna Plendl1 1Department of Veterinary Medicine, Institute of Veterinary Anatomy, Freie Abstract University Berlin, Berlin, Germany The initial intraembryonic vasculogenesis occurs in the cardiogenic mesoderm. Here, 2 College of Veterinary Medicine, School a cell population of proendocardial cells detaches from the mesoderm that subse- of Veterinary and Life Sciences, Murdoch University, Murdoch, WA, Australia quently generates the single endocardial tube by forming vascular plexuses. In the course of embryogenesis, the endocardium retains vasculogenic, angiogenic and Correspondence Johanna Plendl, Department of Veterinary haematopoietic potential. The coronary blood vessels that sustain the rapidly ex- Medicine, Institute of Veterinary Anatomy, panding myocardium develop in the course of the formation of the cardiac loop by Freie University Berlin, Berlin, Germany. Email: [email protected] vasculogenesis and angiogenesis from progenitor cells of the proepicardial serosa at the venous pole of the heart as well as from the endocardium and endothelial cells of Funding information Freie Universität Berlin the sinus venosus. Prospective coronary endothelial cells and progenitor cells of the coronary blood vessel walls (smooth muscle cells, perivascular cells) originate from different cell populations that are in close spatial as well as regulatory connection with each other. Vasculo- and angiogenesis of the coronary blood vessels are for a large part regulated by the epicardium and epicardium-derived cells. Vasculogenic and angiogenic signalling pathways include the vascular endothelial growth factors, the angiopoietins and the fibroblast growth factors and their receptors. -

Cardiovascular System Note: the Cardiovascular System Develops Early (Week 3), Enabling the Embryo to Grow Beyond the Short
Lymphatics: Lymph vessel formation is similar to blood angiogenesis. Lymphatics begin as lymph sacs in three regions: jugular (near brachiocephalic veins); cranial abdominal (future cysterna chyla); and iliac region. Lym- phatic vessels (ducts) form as outgrowths of the sacs. mesenchyme Lymph nodes are produced by localized mesoder- sinusoid lymph duct lumen mal invaginations that partition the vessel lumen into sinu- soids. The mesoderm develops a reticular framework within which mesodermal lymphocytes accumulate. The spleen and hemal nodes (in ruminants) invagination develop similar to the way lymph nodes develop. Lymph Node Formation Prior to birth, fetal circulation is designed for an in utero aqueous environment where the pla- centa oxygenates fetal blood. Suddenly, at birth... Three In-Utero Adjustments ductus Stretching and constriction of arteriosus umbilical arteries shifts fetal blood flow aortic arch from the placenta to the fetus. Reduced pulmonary trunk L atrium venous return through the (left) umbili- foramen ovale R cal vein and ductus venosus allows the atrium latter to gradually close (over a period caudal vena cava of days). Bradykinin released by expand- ductus venosus ing lungs and increased oxygen concen- tration in blood triggers constriction of aorta the ductus arteriosus which, over two liver months, is gradually converted to a fibrous structure, the ligamentum arte- umbilical v. riosum. portal v. The increased blood flow to the lungs and then to the left atrium equalizes pres- sure in the two atria, resulting in closure umbilical aa. of the foramen ovale that eventually grows permanent. 29 The cardiogenic area, the place where the embryonic heart originates, is located . -
Development and Teratology of Cardiovascular and Lymphatic Systems
Development and teratology of cardiovascular and lymphatic systems Repetition: Muscle tissue Beginning of the cardiovascular system development – the 3rd week: Hemangiogenesis (day 15 – 16) – blood islets (insulae sanguinae) in extraembryonic mesoderm and splanchnic mesenchyme of embryo Clusters of mesenchyme cells (angiogenic cells) differentiate into: - angioblasts endothelium (at the periphery of blood islets) - hemoblasts primitive erythrocytes (in the center of blood islets) Clusters of angiogenic cells form a "horseshoe-shaped" space between somatic and splanchnic layer of mesoderm = pericardial cavity. Two endothelial tubes arrise in splanchnic mesoderm. The ventral portion of these tubes forms the cardiogenic area with two heart tubes, while the lateral portions form the dorsal aortae. Germ disc: prosencephalon mesencephalon eye rhombencephalon heart lateral mesoderm somites small blood vessels blood islands 8,9 Spine primitive streak Initially, the cardiogenic area is located anterior to the prechordal plate and the neural plate. The growth of the central nervous system pulls the cardiogenic area and prechordal plate (buccopharyngeal membrane ventrally and caudally ( ). Cardiogenic region just cranial to the prechordal plate. The canalization of cardiogenic clusters in the splanchnic mesoderm results in the formation of the paired heart tubes. Folding of embryo and primitive gut separation from yolk sac. Fusion of the heart tubes a single heart tube is, temporarily attached to the dorsal side of the pericardial cavity by the -

6 Development of the Great Vessels and Conduction Tissue
Development of the Great Vessels and Conduc6on Tissue Development of the heart fields • h:p://php.med.unsw.edu.au/embryology/ index.php?6tle=Advanced_-_Heart_Fields ! 2 Septa6on of the Bulbus Cordis Bulbus Cordis AV Canal Ventricle Looking at a sagital sec6on of the heart early in development the bulbus cordis is con6nuous with the ventricle which is con6nuous with the atria. As the AV canal shiOs to the right the bulbus move to the right as well. Septa6on of the Bulbus Cordis A P A P The next three slides make the point via cross sec6ons that the aorta and pulmonary arteries rotate around each other. This means the septum between them changes posi6on from superior to inferior as well. Septa6on of the Bulbus Cordis P A A P Septa6on of the Bulbus Cordis P A P A Migra6on of neural crest cells Neural crest cells migrate from the 3ed, 4th and 6th pharyngeal arches to form some of the popula6on of cells forming the aor6copulmonary septum. Septa6on of the Bulbus Cordis Truncal (Conal) Swellings Bulbus Cordis The cardiac jelly in the region of the truncus and conus adds the neural crest cells and expands as truncal swellings. Septa6on of the Bulbus Cordis Aorticopulmonary septum These swellings grow toward each other to meet and form the septum between the aorta and pulmonary artery. Aorta Pulmonary Artery Septa6on of the Bulbus Cordis Anterior 1 2 3 1 2 3 The aor6copulmonary septum then rotates as it moves inferiorly. However, the exact mechanism for that rota6on remains unclear. Septa6on of the Bulbus Cordis Aorta Pulmonary Artery Conal Ridges IV Foramen Membranous Muscular IV Endocarial Septum Interventricular Cushion Septum However, the aor6copulmonary septum must form properly for the IV septum to be completed. -

Cardiovascular System Note: the Cardiovascular System Develops Early (Week-3), Enabling the Embryo to Grow Beyond the Short
Cardiovascular System Note: The cardiovascular system develops early (week-3), enabling the embryo to grow beyond the short distances over which diffusion is efficient for transferring 2O , CO2, and cellular nutrients & wastes. Heart: Beginning as a simple tube, the heart undergoes differential growth into a four chambered struc- ture, while it is pumping blood throughout the embryo and into extra-embryonic membranes. Angiogenesis begins with blood island formation in splanchnic mesoderm of the yolk sac and allantois. Vessel formation occurs when island vesicles coalesce, sprout buds, and fuse to form vascular channels. Hematopoiesis (blood cell formation) occurs in the liver and spleen and later in the bone marrow. The transition from fetal to adult circulation involves new vessel formation, vessel merger, and degeneration of early vessels. Formation of a Tubular Heart: The first evidence of heart develop- amnionic cavity ment is bilateral vessel formation within ectoderm the cardiogenic plate (splanchnic meso- embryo derm situated anterior to the embryo). The cardiogenic plate moves ven- tral to the pharynx as the head process cardiogenic yolk sac endoderm mesoderm grows upward and outward. plate Bilateral endocardial tubes meet at the midline & fuse into a single endo- embryo cardial tube, the future heart. Splanchnic mesoderm surround- ing the tube forms cardiac muscle cells heart capable of pumping blood. yolk sac Primitive Heart Regions: Differential growth of the endocardial tube establishes five primitive heart regions: 1] Truncus arteriosus — the output region of the heart. It will develop into the ascending aorta and pulmonary trunk. truncus 2] Bulbus cordis — a bulb-shaped region des- arteriosus tined to become right ventricle. -
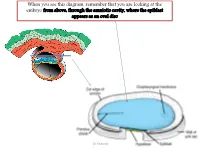
Embryo a the Development of the Heart
When you see this diagram, remember that you are looking at the embryo from above, through the amniotic cavity, where the epiblast appears as an oval disc Dr. Shatarat Dr. Shatarat Why the embryo needs the vascular system? When it appears? Where it appears? with later contributions from neural crest mesenchyme Dr. Shatarat The Cardiac progenitor cells migrate from the Epiblast Through In a Cranial direction on Primitive streak each side of the notochordal process and around the prechordal plate 4 Dr. Shatarat into the splanchnic layer of the lateral plate mesoderm Dr. Shatarat into the splanchnic layer of the lateral plate mesoderm Dr. Shatarat The cells from both sides meet cranially to form the Primary Heart Field (PHF) These cells will form : • The atria • Left ventricle • Part of right ventricle • The remainder of the right ventricle • outflow tract (conus cordis and truncus arteriosus) Are derived from the Secondary Heart Field (SHF) Dr. Shatarat ONE-SOMITE AND TWO-SOMITE STAGES Dr. Shatarat Paired endothelial strands ANGIOBLASTIC CORDS appear in the cardiogenic mesoderm during the third week of development Dr. Shatarat Dr. Shatarat These cords canalize to form two heart tubes that soon fuse as embryo folds laterally to form a single heart tube late in the third week Two hearts tubs The two tubs are Fused Single heart tube is formed As the embryo folds laterally Dr. Shatarat Dr. Shatarat In addition to the cardiogenic region, other blood islands appear bilaterally, parallel and close to the midline of the embryonic shield. These islands form a pair of longitudinal vessels, the dorsal aortae. -

6. Heart and Circulatory System I
6. HEART AND CIRCULATORY SYSTEM I Dr. Taube P. Rothman P&S 12-520 [email protected] 212-305-7930 RECOMMENDED READING: Larsen Human Embryology, 3rd Edition, pp. 195-199; 157-169 top left; 172-174; bottom 181-182; 187-top 189, Simbryo-cardiovascular system SUMMARY: The circulatory system, consisting of heart, blood vessels, and blood cells is the first functional organ to develop. This lecture will focus on the formation of the embryonic vasculature, the origin and formation of the early heart tube and primitive cardiac chambers, cardiac looping, and the primitive circulation. Between the 5th - 8th week of embryonic development, the tubular heart is remodeled into a four chambered structure. We will see how right and left atrioventricular canals connect each atrium with its respective ventricle, and how the atrial septum and definitive right and left atria form. We will also see why the great veins deliver blood to the right atrium while the pulmonary veins empty into the left. GLOSSARY: Angioblasts: precursors of blood vessels Angiogenesis: lengthening, branching, sprouting and remodeling of embryonic blood vessels Aortic arches: paired arteries surrounding the pharynx; portions will contribute to formation of the great arterial vessels Blood islands: clusters of cells in the yolk sac, connecting stalk and chorionic villi that form primitive blood vessels Cardiac jelly: gelatinous extracellular matrix that forms the middle layer of the heart tube Ductus venosus: shunts most of the blood in the umbilical vein into the inferior vena cava -

Cardiovascular System - Accessscience from Mcgraw-Hill Education
Cardiovascular system - AccessScience from McGraw-Hill Education http://accessscience.com/content/109900 (http://accessscience.com/) Article by: Weichert, Charles K. College of Arts and Sciences, University of Cincinnati, Cincinnati, Ohio. Copenhaver, W. M. College of Physicians and Surgeons, Columbia University, New York; Department of Biological Structures, School of Medicine, University of Miami, Miami, Florida. Ebert, James D. Department of Embryology, Carnegie Institution, Washington, DC. Patten, Bradley M. Department of Anatomy, University of Michigan, Ann Arbor, Michigan. Jones, David R. Department of Zoology, University of British Columbia, Vancouver, Canada. Publication year: 2014 DOI: http://dx.doi.org/10.1036/1097-8542.109900 (http://dx.doi.org/10.1036/1097-8542.109900) Content Comparative Anatomy Embryogenesis of blood vessels Balancing ventricular output Heart Angiogenesis Human Postnatal Circulation Arterial system Circulatory system morphogenesis Pulmonary circuit and ductus Venous system Primitive venous system Physiological aspects of transition Comparative Embryology Functional Development of Heart Comparative Physiology Heart Contractions of the heart General physiology of circulation Tubular heart formation Heart-forming areas Microcirculation Cardiac loop and regional development Contractile proteins Heart Formation of definitive heart Synthesis of contractile proteins Arteries Partitioning of mammalian heart Action of inhibitors Venous system Division of atrium and ventricles Human Fetal Circulation at Term Bibliography -

Cardiovascular Development
Cardiovascular Development Handout for “Developmental Biology” Dawei W Dong Learning goals 1. explain the early development of the cardiac tube from visceral mesoderm ahead of the neural plate which is then folded beneath the pharynx of the head fold. 2. outline the fusion of the cardiac tube to form the simple linear heart, the segmentation and the loop formation with sinus venosus, atrium, ventricle, bulbus cordis, and truncus arteriosus. 3. show how septum formation in the primitive heart allows separate pumping of blood into the aortic and the pulmonary trunk. 4. describe the heart inlet separation through the incorporation of the sinus venosus and the pulmonary veins into the atrium. 5. understand the developmental process by which the conus cordis and truncus arteriosus are adapted to give the aortic and pulmonary trunk, i.e., the heart outlet separation. 6. describe the three periods of blood cell formation related to the yolk sac, the liver and the bone marrow. 7. define the three circulatory arcs of the heart to/from the body tissues, the yolk sac (vitelline) and the allantois (umbilical), describe their functions, and understand devel- opmental changes of the arterial and venous systems, in particular, the left and right symmetry breaking. 8. understand the changes of the circulation at birth. Quiz Please test yourself with the quizzes at the end. It does not cover everything, but should give you some hints at how well you have learned the subject. 1 Cardiovascular Development Dawei W Dong 1 Formation and folding of the cardiac tube The cardiac development is specified through BMP signalling originating from the under- lying hypoblast and endoderm.