Bay Annulated Indigo As a New Chromophore for P-Type Dye-Sensitized Solar Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

General Introduction to the Chemistry of Dyes
GENERAL INTRODUCTION TO THE CHEMISTRY OF DYES 1. Principles of Colour Chemistry 1.1 Basis for colour Unlike most organic compounds, dyes possess colour because they 1) absorb light in the visible spectrum (400–700 nm), 2) have at least one chromophore (colour-bearing group), 3) have a conjugated system, i.e. a structure with alternating double and single bonds, and 4) exhibit resonance of electrons, which is a stabilizing force in organic compounds (Abrahart, 1977). When any one of these features is lacking from the molecular structure the colour is lost. In addition to chromophores, most dyes also contain groups known as auxochromes (colour helpers), examples of which are carboxylic acid, sulfonic acid, amino, and hydroxyl groups. While these are not responsible for colour, their presence can shift the colour of a colourant and they are most often used to influence dye solubility. Figure 1 shows the relationships between wavelength of visible and colour absorbed/observed. Other factors contributing to colour are illustrated in Figures 2–4. Fig. 1. Wavelength of light absorption versus colour in organic dyes Wavelength Absorbed (nm) Colour Absorbed Colour Observed 400–435 Violet Yellow-Green 435–480 Blue Yellow 480–490 Green-Blue Orange 490–500 Blue-Green Red 500–560 Green Purple 560–580 Yellow-Green Violet 580–595 Yellow Blue 595–605 Orange Green-Blue 605–700 Red Blue-Green –55– 56 IARC MONOGRAPHS VOLUME 99 Fig. 2. Examples of chromophoric groups present in organic dyes O O N N N N O N N O H N Anthraquinone H N Nitro Azo N N Ar C N N C Ar Methine Phthalocyanine Triarylmethane Fig. -

Identification and Distribution of Dietary Precursors of The
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Vision Research 39 (1999) 219–229 Identification and distribution of dietary precursors of the Drosophila visual pigment chromophore: analysis of carotenoids in wild type and ninaD mutants by HPLC David R. Giovannucci *, Robert S. Stephenson Department of Biological Sciences, Wayne State Uni6ersity, Detroit, MI 48202, USA Received 3 July 1997; received in revised form 10 November 1997 Abstract A dietary source of retinoid or carotenoid has been shown to be necessary for the biosynthesis of functional visual pigment in flies. In the present study, the larvae or adults of Drosophila melanogaster were administered specific carotenoid-containing diets and high performance liquid chromatography was used to identify and quantify the carotenoids in extracts of wild type and ninaD visual mutant flies. When b-carotene was fed to larvae, wild type flies were shown to hydroxylate this molecule and to accumulate zeaxanthin and a small amount of b-cryptoxanthin. Zeaxanthin content was found to increase throughout development and was a major carotenoid peak detected in the adult fly. Carotenoids were twice as effective at mediating zeaxanthin accumulation when provided to larvae versus adults. In the ninaD mutant, zeaxanthin content was shown to be specifically and significantly altered compared to wild type, and was ineffective at mediating visual pigment synthesis when provided to both larval and adult mutant flies. It is proposed that zeaxanthin is the larval storage form for subsequent visual pigment chromophore biosynthesis during pupation, that zeaxanthin or b-crytoxanthin is the immediate precursor for light-independent chromophore synthesis in the adult, and that the ninaD mutant is defective in this pathway. -

Ba3(P1−Xmnxo4)2 : Blue/Green Inorganic Materials Based on Tetrahedral Mn(V)
Bull. Mater. Sci., Vol. 34, No. 6, October 2011, pp. 1257–1262. c Indian Academy of Sciences. Ba3(P1−xMnxO4)2 : Blue/green inorganic materials based on tetrahedral Mn(V) SOURAV LAHA, ROHIT SHARMA, S V BHAT†, MLPREDDY‡, J GOPALAKRISHNAN∗ and S NATARAJAN Solid State and Structural Chemistry Unit, †Department of Physics, Indian Institute of Science, Bangalore 560 012, India ‡Chemical Science and Technology Division, National Institute for Interdisciplinary Science and Technology (NIIST), Thiruvananthapuram 695 019, India MS received 11 May 2011 ) 3− 2 Abstract. We describe a blue/green inorganic material, Ba3(P1−xMnxO4 2 (I) based on tetrahedral MnO4 :3d chromophore. The solid solutions (I) which are sky-blue and turquoise-blue for x ≤ 0·25 and dark green for x ≥ 0·50, 3− are readily synthesized in air from commonly available starting materials, stabilizing the MnO4 chromophore in an isostructural phosphate host. We suggest that the covalency/ionicity of P–O/Mn–O bonds in the solid solutions tunes the crystal field strength around Mn(V) such that a blue colour results for materials with small values of x. The material could serve as a nontoxic blue/green inorganic pigment. Keywords. Blue/green inorganic material; barium phosphate/manganate(V); tetrahedral manganate(V); blue/green chromophore; ligand field tuning of colour. 1. Introduction field transitions within an unusual five coordinated trigo- nal bipyramidal Mn(III). A turquoise-blue solid based on Inorganic solids displaying bright colours are important Li1·33Ti1·66O4 spinel oxide wherein the colour arises from as pigment materials which find a wide range of appli- an intervalence charge-transfer between Ti3+ and Ti4+ has cations in paints, inks, plastics, rubbers, ceramics, ena- also been described recently (Fernández-Osorio et al 2011). -
Color Vision Light
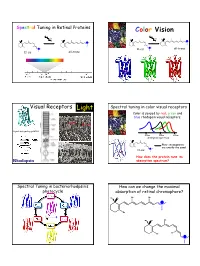
Spectral Tuning in Retinal Proteins Color Vision hν hν N N H H H H N all-trans N 11-cis 11-cis all-trans Visual Receptors Light Spectral tuning in color visual receptors Color is sensed by red, green and blue rhodopsin visual receptors. Rod Cone G-protein signaling pathway 400nm 500nm 600nm absorption spectrum Their chromophores H are exactly the same! 11-cis N How does the protein tune its Rhodopsin absorption spectrum? Spectral Tuning in bacteriorhodpsin’s How can we change the maximal photocycle absorption of retinal chromophore? Me Me Me Me N H Me Me Me Me N H Me Me Me Me Me Me Me H N 1 Excitation energy determines the Electronic Absorption maximal absorption Me Me Me Me π π* N - H S1 Me Absorption of light in the UV-VIS region of the S0 spectrum is due to Response excitation of electrons to higher energy levels. Me Me Me 13 7911 15 N H Me Me π-π* excitation in polyenes π-π* excitation in polyenes π∗ π∗ π∗ π∗ ∆E π∗ E photon E π π π π π Ground state (S0) Excited state (S1) ∆E (excitation energy, band gap) = hν = hc/λ blue-shift red-shift π-π* excitation in polyenes Tuning the length of the conjugated backbone β-carotene O O Vitamin A2 (retinal II) Vitamin A1 (retinal I) Longer wavelength Short wavelength 2 Retinal I Retinal II OPSIN SHIFT: how protein tunes the absorption maximum of its chromophore. Maximal absorption of protonated retinal Schiff base in: Water/methanol solution: 440 nm bR: 568 nm rod Rh: 500 nm red receptor: 560 nm green receptor: 530 nm blue receptor: 426 nm Salmon: different retinals in different stages of life Electrostatics and opsin shift Electrostatics and opsin shift S S positive charge 2 positive charge 2 S2 S2 Me Me Me Me Me Me S1 S1 S1 S1 + + + + N N H H Me O S0 Me O S0 Me O Me O C C Asp (Glu) Asp (Glu) S0 S0 no protein in protein no protein in protein counterion counterion • The counterion stabilizes the positive charge of the chromophore. -

Indigo Chromophores and Pigments Structure and Dynamics
Dyes and Pigments 172 (2020) 107761 Contents lists available at ScienceDirect Dyes and Pigments journal homepage: www.elsevier.com/locate/dyepig Indigo chromophores and pigments: Structure and dynamics T ∗ V.V. Volkova, R. Chellib, R. Righinic, C.C. Perrya, a Interdisciplinary Biomedical Research Centre, School of Science and Technology, Nottingham Trent University, Clifton Lane, Nottingham, NG11 8NS, United Kingdom b Dipartimento di Chimica, Università degli Studi di Firenze, Via della Lastruccia 3, Sesto Fiorentino, 50019, Italy c European Laboratory for Non-Linear Spectroscopy (LENS), Università degli Studi di Firenze, Via Nello Carrara 1, Sesto Fiorentino, 50019, Italy ARTICLE INFO ABSTRACT Keywords: In this study, we explore the molecular mechanisms of the stability of indigo chromophores and pigments. Indigo Assisted with density functional theory, we compare visible, infrared and Raman spectral properties of model Raman molecules, chromophores and pigments derived from living organisms. Using indigo carmine as a representative Infrared model system, we characterize the structure and dynamics of the chromophore in the first electronic excited Density functional theory state using femtosecond visible pump-infrared probe spectroscopy. Results of experiments and theoretical stu- Excited state dies indicate that, while the trans geometry is strongly dominant in the electronic ground state, upon photo- excitation, in the Franck-Condon region, some molecules may experience isomerization and proton transfer dynamics. If this happens, however, the normal modes of the trans geometry of the electronic excited state are reconfirmed within several hundred femtoseconds. Supported by quantum theory, first, we ascribe stabilization of the trans geometry in the Franck-Condon region to the reactive character of the potential energy surface for the indigo chromophore when under the cis geometry in the electronic excited state. -

Molecular Evolution of Color Vision in Vertebrates
Gene 300 (2002) 69–78 www.elsevier.com/locate/gene Molecular evolution of color vision in vertebrates Shozo Yokoyama* Department of Biology, Biological Research Laboratories, Syracuse University, 130 College Place, Syracuse, NY 13244, USA Received 10 December 2001; received in revised form 4 March 2002; accepted 17 July 2002 Abstract Visual systems of vertebrates exhibit a striking level of diversity, reflecting their adaptive responses to various color environments. The photosensitive molecules, visual pigments, can be synthesized in vitro and their absorption spectra can be determined. Comparing the amino acid sequences and absorption spectra of various visual pigments, we can identify amino acid changes that have modified the absorption spectra of visual pigments. These hypotheses can then be tested using the in vitro assay. This approach has been a powerful tool in elucidating not only the molecular bases of color vision, but the processes of adaptive evolution at the molecular level. q 2002 Elsevier Science B.V. All rights reserved. Keywords: Visual pigment; Wavelength absorption; Color vision; Vertebrate 1. Introduction visual pigments, it was necessary to clone opsin genes. This was initiated in 1986 when the opsin genes were cloned Vision has profound effects on the evolution of from cow and human (Nathans and Hogness, 1983, 1984; organisms by affecting survivorship through such behaviors Nathans et al., 1986). The availability of these opsin clones as mating, foraging, and predator avoidance. The data led to the isolation of other orthologous and paralogous collected by vision scientists over the last century genes from various species. Second, in the late 1980s, it demonstrate beyond any doubt that ecology has been a became possible to express opsins in cultured cells, major factor in directing the evolution of visual systems reconstitute with 11-cis-retinal, and measure the absorption (Walls, 1942; Lythgoe, 1979; Jacobs, 1981; Bowmaker, spectra of the resulting visual pigments in vitro (Oprian 1991; Yokoyama and Yokoyama, 1996). -

Chromophore of an Enhanced Green Fluorescent Protein Can Play a Photoprotective Role Due to Photobleaching
International Journal of Molecular Sciences Article Chromophore of an Enhanced Green Fluorescent Protein Can Play a Photoprotective Role Due to Photobleaching Joanna Krasowska 1 , Katarzyna Pierzchała 2,3, Agnieszka Bzowska 1 ,László Forró 3 , Andrzej Sienkiewicz 3,4,5,* and Beata Wielgus-Kutrowska 1,* 1 Division of Biophysics, Institute of Experimental Physics, Faculty of Physics, University of Warsaw, Pasteura 5, 02-093 Warsaw, Poland; [email protected] (J.K.); [email protected] (A.B.) 2 Laboratory for Functional and Metabolic Imaging (LIFMET), Institute of Physics (IPHYS), School of Basic Sciences (SB), Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland; katarzyna.pierzchala@epfl.ch 3 Laboratory of Physics of Complex Matter (LPMC), Institute of Physics (IPHYS), School of Basic Sciences (SB), Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland; laszlo.forro@epfl.ch 4 Laboratory for Quantum Magnetism (LQM), Institute of Physics (IPHYS), School of Basic Sciences (SB), École Polytechnique Fédérale de Lausanne (EPFL), Station 3, CH-1015 Lausanne, Switzerland 5 ADSresonances, Route de Genève 60B, CH-1028 Préverenges, Switzerland * Correspondence: andrzej.sienkiewicz@epfl.ch (A.S.); [email protected] (B.W.-K.) Abstract: Under stress conditions, elevated levels of cellular reactive oxygen species (ROS) may impair crucial cellular structures. To counteract the resulting oxidative damage, living cells are equipped with several defense mechanisms, including photoprotective functions of specific proteins. Here, we discuss the plausible ROS scavenging mechanisms by the enhanced green fluorescent Citation: Krasowska, J.; Pierzchała, protein, EGFP. To check if this protein could fulfill a photoprotective function, we employed electron K.; Bzowska, A.; Forró, L.; Sienkiewicz, A.; Wielgus-Kutrowska, spin resonance (ESR) in combination with spin-trapping. -

THE FLUORESCENCE from the CHROMOPHORE of the PURPLE MEMBRANE PROTEIN Rajni GOVINDJEE, BRIAN BECHER, and THOMAS G
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector THE FLUORESCENCE FROM THE CHROMOPHORE OF THE PURPLE MEMBRANE PROTEIN RAJNi GOVINDJEE, BRIAN BECHER, AND THOMAS G. EBREY, Department ofPhysiology and Biophysics, University ofIllinois, Urbana, Illinois 61801 U.S.A. ABSTRACr The fluorescence from the purple membrane protein (PM) of Halo- bacterium halobium and its relation to the primary photochemical events have been studied. The emission spectrum at 770K has structure, with peaks at 680, 710-715, and 730-735 nm. The excitation spectrum shows a single peak centered at 580 nm. This and a comparison of the fluorescence intensity at 770K under a variety of con- ditions with the amounts of the bathoproduct (or K, the only photoproduct seen at this temperature) formed suggest that the source of the fluorescence is the purple mem- brane itself, not the photoproduct. From the difference in several of their properties, we suggest that the fluorescing state of the pigment is different from the excited state which leads to photoconversion. Light energy absorbed by the purple membrane (PM) of Halobacterium halobium (bacteriorhodopsin) can be converted into chemical free energy of a proton gradient across the cell membrane (Oesterhelt and Stoeckenius, 1973). This pumping of pro- tons across the membrane is associated with the formation and decay of a series of spectroscopic intermediates, the first of which is formed photochemically. The follow- ing is a schematic representation of the cyclic reaction of PM568 (Lozier et al., 1975): PM568 i K628 L M412 --N --0 t hp I In the primary photochemical event, PM hp K, the photon's energy is used to con- vert PM into a high free-energy species, K, whose free energy is used to drive the rest of the cycle (Rosenfeld et al., 1977). -

Shades of Green: Untying the Knots of Green Photoperception
Journal of Experimental Botany, Vol. 71, No. 19 pp. 5764–5770, 2020 doi:10.1093/jxb/eraa312 Advance Access Publication 3 July 2020 This paper is available online free of all access charges (see http://jxb.oxfordjournals.org/open_access.html for further details) REVIEW PAPER Shades of green: untying the knots of green photoperception Martin W. Battle1, , Franco Vegliani2 and Matthew A. Jones2,*, 1 School of Life Sciences, University of Essex, Colchester, UK 2 Institute of Molecular, Cell, and Systems Biology, University of Glasgow, Glasgow, UK * Correspondence: [email protected] Received 24 April 2020; Editorial decision 25 June 2020; Accepted 30 June 2020 Editor: Christine Foyer, University of Birmingham, UK Abstract The development of economical LED technology has enabled the application of different light qualities and quan- tities to control plant growth. Although we have a comprehensive understanding of plants’ perception of red and blue light, the lack of a dedicated green light sensor has frustrated our utilization of intermediate wavelengths, with many contradictory reports in the literature. We discuss the contribution of red and blue photoreceptors to green light perception and highlight how green light can be used to improve crop quality. Importantly, our meta-analysis dem- onstrates that green light perception should instead be considered as a combination of distinct ‘green’ and ‘yellow’ light-induced responses. This distinction will enable clearer interpretation of plants’ behaviour in response to green light as we seek to optimize plant growth and nutritional quality in horticultural contexts. Keywords: Green light, horticulture, LED, photobiology, photoperception, photoreceptor. Introduction: light provides both energy and information to inform plant development Light is a multifaceted signal for plants, providing comprehensive photosynthesis. -

Color Vision in Primates and Other Mammals Carrie C
Teaching Workshop: Color Vision in Primates and Other Mammals Carrie C. Veilleux & Amber Heard-Booth Anthropology Department, University of Texas at Austin Trichromatic Color Vision Trichromatic Color Vision Dichromatic Color Vision Trichromat Dichromat http://www.colblindor.com/coblis-color-blindness-simulator/ How does this work? Dichromats have 2 types of color-detecting cells in the retinas in their eyes: Blue-sensitive cones Yellow/green-sensitive cones Dichromat Green cone - Blue cone How does this work? Trichromats have 3 types of color- detecting cells in the retinas in their eyes: Blue-sensitive cones Yellow/green-sensitive cones Red-sensitive cones Dichromat Trichromat Green cone Red cone - - Green cone Blue cone - Blue cone Most mammals are dichromats Many primates are trichromats Dichromats see a square Trichromats see a square and a circle Dichromatic animals and people can actually detect camouflaged objects better than trichromats 73 5 45 Physiology of Color Vision Cone photoreceptors Physiology of Color Vision Photopigment Opsin protein chromophore chromophore (11-cis retinal) opsin protein Physiology of Color Vision (Fasick et al. 2002) Cone spectral sensitivity (color of light to which it responds) is determined by structure of opsin protein (amino acid sequence of protein). Amino acid sequence is determined by opsin genes. Physiology of Color Vision So, changes in the DNA sequence of an opsin gene can lead to differences in the spectral sensitivity of cones (“critical tuning sites”). critical tuning sites of the M/L opsin gene Nathans (1999) Physiology of Color Vision So, changes in the DNA sequence of an opsin gene can lead to differences in the spectral sensitivity of cones (“critical tuning sites”). -

Color Detection Using Chromophore-Nanotube Hybrid
Color Detection Using Chromophore-Nanotube Hybrid Devices Xinjian Zhou*, Thomas Zifer, Bryan M. Wong, Karen L. Krafcik, François Léonard and Andrew L. Vance Sandia National Laboratories, Livermore, CA, 94551 *Corresponding author. E-mail: [email protected]. ABSTRACT We present a nanoscale color detector based on a single-walled carbon nanotube functionalized with azobenzene chromophores, where the chromophores serve as photoabsorbers and the nanotube as the electronic read-out. By synthesizing chromophores with specific absorption windows in the visible spectrum and anchoring them to the nanotube surface, we demonstrate the controlled detection of visible light of low intensity in narrow ranges of wavelengths. Our measurements suggest that upon photoabsorption, the chromophores isomerize from the ground state trans configuration to the excited state cis configuration, accompanied by a large change in dipole moment, changing the electrostatic environment of the nanotube. All-electron ab initio calculations are used to study the chromophore-nanotube hybrids, and show that the chromophores bind strongly to the nanotubes without disturbing the electronic structure of either 1 species. Calculated values of the dipole moments support the notion of dipole changes as the optical detection mechanism. 2 Phototransduction plays a central role in key natural processes such as vision and photosynthesis. For these processes, natural evolution has found exquisite solutions that optimize efficiency, compactness, and self-healing. While researchers have long tried to supplant nature’s successes using solid-state approaches, our ability to convert optical radiation to electrical signals with nanoscale precision still remains in its infancy 1-4. An alternative to solid-state approaches is one that more closely mimics natural processes by integrating chemical or biological materials with solid-state materials. -

Photo-Isomerization Upshifts the Pka of the Photoactive Yellow Protein
Journal of Photochemistry and Photobiology A: Chemistry 270 (2013) 43–52 Contents lists available at ScienceDirect Journal of Photochemistry and Photobiology A: Chemistry journal homepage: www.elsevier.com/locate/jphotochem Photo-isomerization upshifts the pKa of the Photoactive Yellow Protein chromophore to contribute to photocycle propagation a,1 b,1 a d Sadia Naseem , Adèle D. Laurent , Elizabeth C. Carroll , Mikas Vengris , c c b a,∗ Masato Kumauchi , Wouter D. Hoff , Anna I. Krylov , Delmar S. Larsen a Department of Chemistry, University of California, Davis, One Shields Avenue, Davis, CA 95616 USA b Department of Chemistry, University of Southern California, Los Angeles, CA 90089-0482, USA c Department of Microbiology & Molecular Genetics, Oklahoma State University, Stillwater, OK 74078, USA d Quantum Electronics Department, Faculty of Physics, Vilnius University Sauletekio 10, LT10223 Vilnius, Lithuania a r a t i b s c l t r e a i n f o c t Article history: The influence of chromophore structure on the protonation constant of the Photoactive Yellow Protein Received 11 March 2012 chromophore is explored with isolated para-coumaric acid (pCA) and thiomethyl-para-coumaric acid Received in revised form 19 June 2013 model chromophores in solution. pH titration coupled with visible absorption spectra of the trans and Accepted 25 June 2013 photogenerated cis conformer of isolated pCA demonstrates that the isomerization of the chromophore Available online 9 July 2013 increases the pKa of the phenolate group by 0.6 units (to 10.1 ± 0.22). Formation of the pCA thioester reduces the pKa of the phenolic group by 0.3 units (from 9.5 ± 0.15 to 9.2 ± 0.16).