Supercritical Fluid Chromatography
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lecture Notes: BCS Theory of Superconductivity
Lecture Notes: BCS theory of superconductivity Prof. Rafael M. Fernandes Here we will discuss a new ground state of the interacting electron gas: the superconducting state. In this macroscopic quantum state, the electrons form coherent bound states called Cooper pairs, which dramatically change the macroscopic properties of the system, giving rise to perfect conductivity and perfect diamagnetism. We will mostly focus on conventional superconductors, where the Cooper pairs originate from a small attractive electron-electron interaction mediated by phonons. However, in the so- called unconventional superconductors - a topic of intense research in current solid state physics - the pairing can originate even from purely repulsive interactions. 1 Phenomenology Superconductivity was discovered by Kamerlingh-Onnes in 1911, when he was studying the transport properties of Hg (mercury) at low temperatures. He found that below the liquifying temperature of helium, at around 4:2 K, the resistivity of Hg would suddenly drop to zero. Although at the time there was not a well established model for the low-temperature behavior of transport in metals, the result was quite surprising, as the expectations were that the resistivity would either go to zero or diverge at T = 0, but not vanish at a finite temperature. In a metal the resistivity at low temperatures has a constant contribution from impurity scattering, a T 2 contribution from electron-electron scattering, and a T 5 contribution from phonon scattering. Thus, the vanishing of the resistivity at low temperatures is a clear indication of a new ground state. Another key property of the superconductor was discovered in 1933 by Meissner. -

Supercritical Fluid Extraction of Positron-Emitting Radioisotopes from Solid Target Matrices
XA0101188 11. United States of America Supercritical Fluid Extraction of Positron-Emitting Radioisotopes From Solid Target Matrices D. Schlyer, Brookhaven National Laboratory, Chemistry Department, Upton, Bldg. 901, New York 11973-5000, USA Project Description Supercritical fluids are attractive as media for both chemical reactions, as well as process extraction since their physical properties can be manipulated by small changes in pressure and temperature near the critical point of the fluid. What is a supercritical fluid? Above a certain temperature, a vapor can no longer be liquefied regardless of pressure critical temperature - Tc supercritical fluid r«gi on solid a u & temperature Fig. 1. Phase diagram depicting regions of solid, liquid, gas and supercritical fluid behavior. The critical point is defined by a critical pressure (Pc) and critical temperature (Tc) for a particular substance. Such changes can result in drastic effects on density-dependent properties such as solubility, refractive index, dielectric constant, viscosity and diffusivity of the fluid[l,2,3]. This suggests that pressure tuning of a pure supercritical fluid may be a useful means to manipulate chemical reactions on the basis of a thermodynamic solvent effect. It also means that the solvation properties of the fluid can be precisely controlled to enable selective component extraction from a matrix. In recent years there has been a growing interest in applying supercritical fluid extraction to the selective removal of trace metals from solid samples [4-10]. Much of the work has been done on simple systems comprised of inert matrices such as silica or cellulose. Recently, this process as been expanded to environmental samples as well [11,12]. -

General Introduction to the Chemistry of Dyes
GENERAL INTRODUCTION TO THE CHEMISTRY OF DYES 1. Principles of Colour Chemistry 1.1 Basis for colour Unlike most organic compounds, dyes possess colour because they 1) absorb light in the visible spectrum (400–700 nm), 2) have at least one chromophore (colour-bearing group), 3) have a conjugated system, i.e. a structure with alternating double and single bonds, and 4) exhibit resonance of electrons, which is a stabilizing force in organic compounds (Abrahart, 1977). When any one of these features is lacking from the molecular structure the colour is lost. In addition to chromophores, most dyes also contain groups known as auxochromes (colour helpers), examples of which are carboxylic acid, sulfonic acid, amino, and hydroxyl groups. While these are not responsible for colour, their presence can shift the colour of a colourant and they are most often used to influence dye solubility. Figure 1 shows the relationships between wavelength of visible and colour absorbed/observed. Other factors contributing to colour are illustrated in Figures 2–4. Fig. 1. Wavelength of light absorption versus colour in organic dyes Wavelength Absorbed (nm) Colour Absorbed Colour Observed 400–435 Violet Yellow-Green 435–480 Blue Yellow 480–490 Green-Blue Orange 490–500 Blue-Green Red 500–560 Green Purple 560–580 Yellow-Green Violet 580–595 Yellow Blue 595–605 Orange Green-Blue 605–700 Red Blue-Green –55– 56 IARC MONOGRAPHS VOLUME 99 Fig. 2. Examples of chromophoric groups present in organic dyes O O N N N N O N N O H N Anthraquinone H N Nitro Azo N N Ar C N N C Ar Methine Phthalocyanine Triarylmethane Fig. -

Equation of State and Phase Transitions in the Nuclear
National Academy of Sciences of Ukraine Bogolyubov Institute for Theoretical Physics Has the rights of a manuscript Bugaev Kyrill Alekseevich UDC: 532.51; 533.77; 539.125/126; 544.586.6 Equation of State and Phase Transitions in the Nuclear and Hadronic Systems Speciality 01.04.02 - theoretical physics DISSERTATION to receive a scientific degree of the Doctor of Science in physics and mathematics arXiv:1012.3400v1 [nucl-th] 15 Dec 2010 Kiev - 2009 2 Abstract An investigation of strongly interacting matter equation of state remains one of the major tasks of modern high energy nuclear physics for almost a quarter of century. The present work is my doctor of science thesis which contains my contribution (42 works) to this field made between 1993 and 2008. Inhere I mainly discuss the common physical and mathematical features of several exactly solvable statistical models which describe the nuclear liquid-gas phase transition and the deconfinement phase transition. Luckily, in some cases it was possible to rigorously extend the solutions found in thermodynamic limit to finite volumes and to formulate the finite volume analogs of phases directly from the grand canonical partition. It turns out that finite volume (surface) of a system generates also the temporal constraints, i.e. the finite formation/decay time of possible states in this finite system. Among other results I would like to mention the calculation of upper and lower bounds for the surface entropy of physical clusters within the Hills and Dales model; evaluation of the second virial coefficient which accounts for the Lorentz contraction of the hard core repulsing potential between hadrons; inclusion of large width of heavy quark-gluon bags into statistical description. -

Identification and Distribution of Dietary Precursors of The
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Vision Research 39 (1999) 219–229 Identification and distribution of dietary precursors of the Drosophila visual pigment chromophore: analysis of carotenoids in wild type and ninaD mutants by HPLC David R. Giovannucci *, Robert S. Stephenson Department of Biological Sciences, Wayne State Uni6ersity, Detroit, MI 48202, USA Received 3 July 1997; received in revised form 10 November 1997 Abstract A dietary source of retinoid or carotenoid has been shown to be necessary for the biosynthesis of functional visual pigment in flies. In the present study, the larvae or adults of Drosophila melanogaster were administered specific carotenoid-containing diets and high performance liquid chromatography was used to identify and quantify the carotenoids in extracts of wild type and ninaD visual mutant flies. When b-carotene was fed to larvae, wild type flies were shown to hydroxylate this molecule and to accumulate zeaxanthin and a small amount of b-cryptoxanthin. Zeaxanthin content was found to increase throughout development and was a major carotenoid peak detected in the adult fly. Carotenoids were twice as effective at mediating zeaxanthin accumulation when provided to larvae versus adults. In the ninaD mutant, zeaxanthin content was shown to be specifically and significantly altered compared to wild type, and was ineffective at mediating visual pigment synthesis when provided to both larval and adult mutant flies. It is proposed that zeaxanthin is the larval storage form for subsequent visual pigment chromophore biosynthesis during pupation, that zeaxanthin or b-crytoxanthin is the immediate precursor for light-independent chromophore synthesis in the adult, and that the ninaD mutant is defective in this pathway. -

Ba3(P1−Xmnxo4)2 : Blue/Green Inorganic Materials Based on Tetrahedral Mn(V)
Bull. Mater. Sci., Vol. 34, No. 6, October 2011, pp. 1257–1262. c Indian Academy of Sciences. Ba3(P1−xMnxO4)2 : Blue/green inorganic materials based on tetrahedral Mn(V) SOURAV LAHA, ROHIT SHARMA, S V BHAT†, MLPREDDY‡, J GOPALAKRISHNAN∗ and S NATARAJAN Solid State and Structural Chemistry Unit, †Department of Physics, Indian Institute of Science, Bangalore 560 012, India ‡Chemical Science and Technology Division, National Institute for Interdisciplinary Science and Technology (NIIST), Thiruvananthapuram 695 019, India MS received 11 May 2011 ) 3− 2 Abstract. We describe a blue/green inorganic material, Ba3(P1−xMnxO4 2 (I) based on tetrahedral MnO4 :3d chromophore. The solid solutions (I) which are sky-blue and turquoise-blue for x ≤ 0·25 and dark green for x ≥ 0·50, 3− are readily synthesized in air from commonly available starting materials, stabilizing the MnO4 chromophore in an isostructural phosphate host. We suggest that the covalency/ionicity of P–O/Mn–O bonds in the solid solutions tunes the crystal field strength around Mn(V) such that a blue colour results for materials with small values of x. The material could serve as a nontoxic blue/green inorganic pigment. Keywords. Blue/green inorganic material; barium phosphate/manganate(V); tetrahedral manganate(V); blue/green chromophore; ligand field tuning of colour. 1. Introduction field transitions within an unusual five coordinated trigo- nal bipyramidal Mn(III). A turquoise-blue solid based on Inorganic solids displaying bright colours are important Li1·33Ti1·66O4 spinel oxide wherein the colour arises from as pigment materials which find a wide range of appli- an intervalence charge-transfer between Ti3+ and Ti4+ has cations in paints, inks, plastics, rubbers, ceramics, ena- also been described recently (Fernández-Osorio et al 2011). -
Color Vision Light
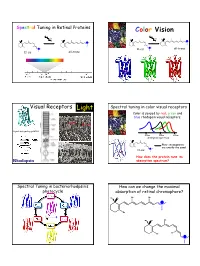
Spectral Tuning in Retinal Proteins Color Vision hν hν N N H H H H N all-trans N 11-cis 11-cis all-trans Visual Receptors Light Spectral tuning in color visual receptors Color is sensed by red, green and blue rhodopsin visual receptors. Rod Cone G-protein signaling pathway 400nm 500nm 600nm absorption spectrum Their chromophores H are exactly the same! 11-cis N How does the protein tune its Rhodopsin absorption spectrum? Spectral Tuning in bacteriorhodpsin’s How can we change the maximal photocycle absorption of retinal chromophore? Me Me Me Me N H Me Me Me Me N H Me Me Me Me Me Me Me H N 1 Excitation energy determines the Electronic Absorption maximal absorption Me Me Me Me π π* N - H S1 Me Absorption of light in the UV-VIS region of the S0 spectrum is due to Response excitation of electrons to higher energy levels. Me Me Me 13 7911 15 N H Me Me π-π* excitation in polyenes π-π* excitation in polyenes π∗ π∗ π∗ π∗ ∆E π∗ E photon E π π π π π Ground state (S0) Excited state (S1) ∆E (excitation energy, band gap) = hν = hc/λ blue-shift red-shift π-π* excitation in polyenes Tuning the length of the conjugated backbone β-carotene O O Vitamin A2 (retinal II) Vitamin A1 (retinal I) Longer wavelength Short wavelength 2 Retinal I Retinal II OPSIN SHIFT: how protein tunes the absorption maximum of its chromophore. Maximal absorption of protonated retinal Schiff base in: Water/methanol solution: 440 nm bR: 568 nm rod Rh: 500 nm red receptor: 560 nm green receptor: 530 nm blue receptor: 426 nm Salmon: different retinals in different stages of life Electrostatics and opsin shift Electrostatics and opsin shift S S positive charge 2 positive charge 2 S2 S2 Me Me Me Me Me Me S1 S1 S1 S1 + + + + N N H H Me O S0 Me O S0 Me O Me O C C Asp (Glu) Asp (Glu) S0 S0 no protein in protein no protein in protein counterion counterion • The counterion stabilizes the positive charge of the chromophore. -

Lecture 5 Non-Aqueous Phase Liquid (NAPL) Fate and Transport
Lecture 5 Non-Aqueous Phase Liquid (NAPL) Fate and Transport Factors affecting NAPL movement Fluid properties: Porous medium: 9 Density Permeability 9 Interfacial tension Pore size 9 Residual saturation Structure Partitioning properties Solubility Ground water: Volatility and vapor density Water content 9 Velocity Partitioning processes NAPL can partition between four phases: NAPL Gas Solid (vapor) Aqueous solution Water to gas partitioning (volatilization) Aqueous ↔ gaseous Henry’s Law (for dilute solutions) 3 Dimensionless (CG, CW in moles/m ) C G = H′ CW Dimensional (P = partial pressure in atm) P = H CW Henry’s Law Constant H has dimensions: atm m3 / mol H’ is dimensionless H’ = H/RT R = gas constant = 8.20575 x 10-5 atm m3/mol °K T = temperature in °K NAPL to gas partitioning (volatilization) NAPL ↔ gaseous Raoult’s Law: CG = Xt (P°/RT) Xt = mole fraction of compound in NAPL [-] P° = pure compound vapor pressure [atm] R = universal gas constant [m3-atm/mole/°K] T = temperature [°K] Volatility Vapor pressure P° is measure of volatility P° > 1.3 x 10-3 atm → compound is “volatile” 1.3 x 10-3 > P° > 1.3 x 10-13 atm → compound is “semi-volatile” Example: equilibrium with benzene P° = 76 mm Hg at 20°C = 0.1 atm R = 8.205 x 10-5 m3-atm/mol/°K T = 20°C (assumed) = 293°K Assume 100% benzene, mole fraction Xt = 1 3 CG = Xt P°/(RT) = 4.16 mol/m Molecular weight of benzene, C6H6 = 78 g/mol 3 3 CG = 4.16 mol/m × 78 g/mol = 324 g/m = 0.32 g/L 6 CG = 0.32 g/L x 24 L/mol / (78 g/mol) x 10 = 99,000 ppmv One mole of ideal gas = 22.4 L at STP (1 atm, 0 C), Corrected to 20 C: 293/273*22.4 = 24.0 L/mol Gas concentration in equilibrium with pure benzene NAPL Example: equilibrium with gasoline Gasoline is complex mixture – mole fraction is difficult to determine and varies Benzene = 1 to several percent (Cline et al., 1991) Based on analysis reported by Johnson et al. -

Introduction to Unconventional Superconductivity Manfred Sigrist
Introduction to Unconventional Superconductivity Manfred Sigrist Theoretische Physik, ETH-Hönggerberg, 8093 Zürich, Switzerland Abstract. This lecture gives a basic introduction into some aspects of the unconventionalsupercon- ductivity. First we analyze the conditions to realized unconventional superconductivity in strongly correlated electron systems. Then an introduction of the generalized BCS theory is given and sev- eral key properties of unconventional pairing states are discussed. The phenomenological treatment based on the Ginzburg-Landau formulations provides a view on unconventional superconductivity based on the conceptof symmetry breaking.Finally some aspects of two examples will be discussed: high-temperature superconductivity and spin-triplet superconductivity in Sr2RuO4. Keywords: Unconventional superconductivity, high-temperature superconductivity, Sr2RuO4 INTRODUCTION Superconductivity remains to be one of the most fascinating and intriguing phases of matter even nearly hundred years after its first observation. Owing to the breakthrough in 1957 by Bardeen, Cooper and Schrieffer we understand superconductivity as a conden- sate of electron pairs, so-called Cooper pairs, which form due to an attractive interaction among electrons. In the superconducting materials known until the mid-seventies this interaction is mediated by electron-phonon coupling which gises rise to Cooper pairs in the most symmetric form, i.e. vanishing relative orbital angular momentum and spin sin- glet configuration (nowadays called s-wave pairing). After the introduction of the BCS concept, also studies of alternative pairing forms started. Early on Anderson and Morel [1] as well as Balian and Werthamer [2] investigated superconducting phases which later would be identified as the A- and the B-phase of superfluid 3He [3]. In contrast to the s-wave superconductors the A- and B-phase are characterized by Cooper pairs with an- gular momentum 1 and spin-triplet configuration. -

Phase Diagrams a Phase Diagram Is Used to Show the Relationship Between Temperature, Pressure and State of Matter
Phase Diagrams A phase diagram is used to show the relationship between temperature, pressure and state of matter. Before moving ahead, let us review some vocabulary and particle diagrams. States of Matter Solid: rigid, has definite volume and definite shape Liquid: flows, has definite volume, but takes the shape of the container Gas: flows, no definite volume or shape, shape and volume are determined by container Plasma: atoms are separated into nuclei (neutrons and protons) and electrons, no definite volume or shape Changes of States of Matter Freezing start as a liquid, end as a solid, slowing particle motion, forming more intermolecular bonds Melting start as a solid, end as a liquid, increasing particle motion, break some intermolecular bonds Condensation start as a gas, end as a liquid, decreasing particle motion, form intermolecular bonds Evaporation/Boiling/Vaporization start as a liquid, end as a gas, increasing particle motion, break intermolecular bonds Sublimation Starts as a solid, ends as a gas, increases particle speed, breaks intermolecular bonds Deposition Starts as a gas, ends as a solid, decreases particle speed, forms intermolecular bonds http://phet.colorado.edu/en/simulation/states- of-matter The flat sections on the graph are the points where a phase change is occurring. Both states of matter are present at the same time. In the flat sections, heat is being removed by the formation of intermolecular bonds. The flat points are phase changes. The heat added to the system are being used to break intermolecular bonds. PHASE DIAGRAMS Phase diagrams are used to show when a specific substance will change its state of matter (alignment of particles and distance between particles). -

Indigo Chromophores and Pigments Structure and Dynamics
Dyes and Pigments 172 (2020) 107761 Contents lists available at ScienceDirect Dyes and Pigments journal homepage: www.elsevier.com/locate/dyepig Indigo chromophores and pigments: Structure and dynamics T ∗ V.V. Volkova, R. Chellib, R. Righinic, C.C. Perrya, a Interdisciplinary Biomedical Research Centre, School of Science and Technology, Nottingham Trent University, Clifton Lane, Nottingham, NG11 8NS, United Kingdom b Dipartimento di Chimica, Università degli Studi di Firenze, Via della Lastruccia 3, Sesto Fiorentino, 50019, Italy c European Laboratory for Non-Linear Spectroscopy (LENS), Università degli Studi di Firenze, Via Nello Carrara 1, Sesto Fiorentino, 50019, Italy ARTICLE INFO ABSTRACT Keywords: In this study, we explore the molecular mechanisms of the stability of indigo chromophores and pigments. Indigo Assisted with density functional theory, we compare visible, infrared and Raman spectral properties of model Raman molecules, chromophores and pigments derived from living organisms. Using indigo carmine as a representative Infrared model system, we characterize the structure and dynamics of the chromophore in the first electronic excited Density functional theory state using femtosecond visible pump-infrared probe spectroscopy. Results of experiments and theoretical stu- Excited state dies indicate that, while the trans geometry is strongly dominant in the electronic ground state, upon photo- excitation, in the Franck-Condon region, some molecules may experience isomerization and proton transfer dynamics. If this happens, however, the normal modes of the trans geometry of the electronic excited state are reconfirmed within several hundred femtoseconds. Supported by quantum theory, first, we ascribe stabilization of the trans geometry in the Franck-Condon region to the reactive character of the potential energy surface for the indigo chromophore when under the cis geometry in the electronic excited state. -

States of Matter
States of Matter A Museum of Science Traveling Program Description States of Matter is a 60- minute presentation about the characteristics of solids, liquids, gases, and the temperature- dependent transitions between them. It is designed to build on NGSS-based curricula. NGSS: Next Generation Science Standards Needs We bring all materials and equipment, including a video projector and screen. Access to 110- volt electricity is required. Space Requirements The program can be presented in assembly- suitable spaces like gyms, multipurpose rooms, cafeterias, and auditoriums. Goals: Changing State A major goal is to show that all substances will go through phase changes and that every substance has its own temperatures at which it changes state. Goals: Boiling Liquid nitrogen boils when exposed to room temperature. When used with every day objects, it can lead to unexpected results! Goals: Condensing Placing balloons in liquid nitrogen causes the gas inside to condense, shrinking the balloons before the eyes of the audience! Latex-free balloons are available for schools with allergic students. Goals: Freezing Nitrogen can also be used to flash-freeze a variety of liquids. Students are encouraged to predict the results, and may end up surprised! Goals: Behavior Changes When solids encounter liquid nitrogen, they can no longer change state, but their properties may have drastic—sometimes even shattering— changes. Finale Likewise, the properties of gas can change at extreme temperatures. At the end of the program, a blast of steam is superheated… Finale …incinerating a piece of paper! Additional Content In addition to these core goals, other concepts are taught with a variety of additional demonstrations.