Indigo Chromophores and Pigments Structure and Dynamics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

General Introduction to the Chemistry of Dyes
GENERAL INTRODUCTION TO THE CHEMISTRY OF DYES 1. Principles of Colour Chemistry 1.1 Basis for colour Unlike most organic compounds, dyes possess colour because they 1) absorb light in the visible spectrum (400–700 nm), 2) have at least one chromophore (colour-bearing group), 3) have a conjugated system, i.e. a structure with alternating double and single bonds, and 4) exhibit resonance of electrons, which is a stabilizing force in organic compounds (Abrahart, 1977). When any one of these features is lacking from the molecular structure the colour is lost. In addition to chromophores, most dyes also contain groups known as auxochromes (colour helpers), examples of which are carboxylic acid, sulfonic acid, amino, and hydroxyl groups. While these are not responsible for colour, their presence can shift the colour of a colourant and they are most often used to influence dye solubility. Figure 1 shows the relationships between wavelength of visible and colour absorbed/observed. Other factors contributing to colour are illustrated in Figures 2–4. Fig. 1. Wavelength of light absorption versus colour in organic dyes Wavelength Absorbed (nm) Colour Absorbed Colour Observed 400–435 Violet Yellow-Green 435–480 Blue Yellow 480–490 Green-Blue Orange 490–500 Blue-Green Red 500–560 Green Purple 560–580 Yellow-Green Violet 580–595 Yellow Blue 595–605 Orange Green-Blue 605–700 Red Blue-Green –55– 56 IARC MONOGRAPHS VOLUME 99 Fig. 2. Examples of chromophoric groups present in organic dyes O O N N N N O N N O H N Anthraquinone H N Nitro Azo N N Ar C N N C Ar Methine Phthalocyanine Triarylmethane Fig. -

Indigo Fructose Dye Vat
Kraftkolour Pty Ltd Factory 2, 99 Heyington Ave THOMASTOWN Vic 3074 Tel: 1300 720 493 Web: Kraftkolour.net.au Indigo Fructose Dye Vat The fructose indigo vat was developed by Michel Garcia. The addition of the fructose sugar acts as a reducing agent to the Indigo. The sugar removes one of the oxygen molecules from the indigo making it soluble in water. The addition of the Calcium Hydroxide (slaked or hydrated lime) changes the pH from an acid to a base. The proper pH to get good colour on wool should be about +9 and for cotton and cellulose +10. When the yarn or fabric is dipped into the indigo dye vat, it turns a green colour. When the yarn is raised into the air, the oxygen molecules from the air, bind with the indigo and turn the green into blue. To get darker and more intense blues, the yarn needs to be dipped into the indigo vat and raised into the air to oxidize several times. The colour builds up onto the yarn or cloth in layers. Keep dipping and airing out the yarn until the desired level of colour is achieved. An Indigo vat can be re-used and kept alive for several weeks until all of the indigo has been exhausted. If the Vat still has indigo but has turned blue, reheat the Vat to 50 deg C. Check the pH. Add about a teaspoon of fructose crystals and wait 15-30 minutes. The Vat should turn green. If it is still blue add some Calcium Hydroxide. -

Tyrian Purple (6,6'-Dibromoindigo)
Tyrian Purple (6,6’-dibromoindigo) A New Twist on the Dye of Old An Ancient Process The Current Way The base chemical of 6,6’-dibromoindigo dye is Dow Chemical Company was the first company to make found naturally in mollusks and certain other crustaceans. Fabric can be dyed through synthetic indigo dye. 6,6’-dibromoindigo soon followed. Indigo direct dyeing, where the fabric or fiber is dyes (including 6,6’-dibromoindigo) are no longer made in the coated with the paste of the mollusk’s mucus U.S., because it is cheaper to import them from other countries. gland. The pasted fabric is allowed to sit in the Today, indigo dye is produced using laboratory chemical sun so that the purple can develop. Fabric can processes. These processes are highly efficient and cost-effective also be dyed using vat dyeing. In this process, the saliva of the mollusk is combined with for the companies that use them, but there are a growing paste of the mucus gland and allowed to dry. number of environmental concerns that are associated with their This residue is ground into a powder and put manufacture and use, as the dying process creates a large into a warm solution of sodium hydroxide or amount of chemical waste that must be disposed of carefully. At lye, away from sunlight, and fabric this time, both lawmakers and chemists are investigating simpler, is immersed in it. Finally, the dyed fabric is put through a finishing process (for safer, and more efficient ways to get the vividly colored clothing example, an acid wash), and washed with soap and water. -

H'mong Ancient Methods of Indigo Dyeing and Beeswax Batik in Cat
International Journal of Science and Research (IJSR) ISSN: 2319-7064 ResearchGate Impact Factor (2018): 0.28 | SJIF (2019): 7.583 H’mong Ancient Methods of Indigo Dyeing and Beeswax Batik in Cat CAT Village, Hoang Lien Commune, SAPA Town, Lao Cai Province, Vietnam Le Thi Hanh Lien1*, Nguyen Thi Hai Yen2, Dao Thi Luu3, Phi Thi Thu Hoang4, Nguyen Thuy Linh5 1, 2, 3, 4 Institute of Geography, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet Road, Nghia Do, CauGiay District, Ha Noi, Vietnam 5Management Development Institute of Singapore *Corresponding author: lehanhlien2017[at]gmail.com Abstract: Indigo dyeing and beeswax batik are the two traditional crafts that have long been associated with the H’Mong people in Sa Pa in general, Cat Cat village in particular and still preserved until the presentday. Through many stages of making indigo dye combined with sophisticated techniques and the ingenuity, meticulousness of the artisans in each motif and pattern, unique beeswax batik and indigo dyeing products have been created bringing the cultural identity of the H’mong people. These handicraft products have become a highlight to attract tourists to learn and discover local cultural values and they are meaningful souvenirs for visitors after each trip. In recent years, the development of the community-based tourism model in Cat Cat village has brought many benefits to the local community. Meanwhile, it has also contributed to creating opportunities for the development and restoration of H’mong traditional crafts. Keywords: indigo dyeing, beeswax batik, H’Mong, Cat Cat, Sa Pa 1. Introduction cultural and religious life of the H'Mong. -

Identification and Distribution of Dietary Precursors of The
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Vision Research 39 (1999) 219–229 Identification and distribution of dietary precursors of the Drosophila visual pigment chromophore: analysis of carotenoids in wild type and ninaD mutants by HPLC David R. Giovannucci *, Robert S. Stephenson Department of Biological Sciences, Wayne State Uni6ersity, Detroit, MI 48202, USA Received 3 July 1997; received in revised form 10 November 1997 Abstract A dietary source of retinoid or carotenoid has been shown to be necessary for the biosynthesis of functional visual pigment in flies. In the present study, the larvae or adults of Drosophila melanogaster were administered specific carotenoid-containing diets and high performance liquid chromatography was used to identify and quantify the carotenoids in extracts of wild type and ninaD visual mutant flies. When b-carotene was fed to larvae, wild type flies were shown to hydroxylate this molecule and to accumulate zeaxanthin and a small amount of b-cryptoxanthin. Zeaxanthin content was found to increase throughout development and was a major carotenoid peak detected in the adult fly. Carotenoids were twice as effective at mediating zeaxanthin accumulation when provided to larvae versus adults. In the ninaD mutant, zeaxanthin content was shown to be specifically and significantly altered compared to wild type, and was ineffective at mediating visual pigment synthesis when provided to both larval and adult mutant flies. It is proposed that zeaxanthin is the larval storage form for subsequent visual pigment chromophore biosynthesis during pupation, that zeaxanthin or b-crytoxanthin is the immediate precursor for light-independent chromophore synthesis in the adult, and that the ninaD mutant is defective in this pathway. -

Roadmap of Solid-State Lithium-Organic Batteries Toward 500 Wh Kg−1 † † Lihong Zhao, Alae Eddine Lakraychi, Zhaoyang Chen, Yanliang Liang, and Yan Yao*
Focus Review http://pubs.acs.org/journal/aelccp Roadmap of Solid-State Lithium-Organic Batteries toward 500 Wh kg−1 † † Lihong Zhao, Alae Eddine Lakraychi, Zhaoyang Chen, Yanliang Liang, and Yan Yao* Cite This: ACS Energy Lett. 2021, 6, 3287−3306 Read Online ACCESS Metrics & More Article Recommendations *sı Supporting Information ABSTRACT: Over the past few years, solid-state electrolytes (SSEs) have attracted tremendous attention due to their credible promise toward high-energy batteries. In parallel, organic battery electrode materials (OBEMs) are gaining momentum as strong candidates thanks to their lower environmental footprint, flexibility in molecular design and high energy metrics. Integration of the two constitutes a potential synergy to enable energy-dense solid- state batteries (SSBs) with high safety, low cost, and long-term sustainability. In this Review, we present the technological feasibility of combining OBEMs with SSEs along with the possible cell configurations that may result from this peculiar combination. We provide an overview of organic SSBs and discuss their main challenges. We analyze the performance-limiting factors and the critical cell design parameters governing cell-level specific energy and energy density. Lastly, we propose guidelines to achieve 500 Wh kg−1 cell-level specific energy with solid-state Li−organic batteries. Downloaded via Yan Yao on August 25, 2021 at 16:56:41 (UTC). rganic battery electrode materials (OBEMs) have molecules (<2 g cm−3) penalizes the energy density received considerable attention in the past few years. (volumetric) of assembled cells; second, low electronic O With a chemical composition derived from naturally conductivity imposes the use of large amount of conductive abundant elements (C, H, N, O, and S), a real possibility of agents which lower the cell-level specific energy (gravimetric); being generated from renewable resources (biomass), and an See https://pubs.acs.org/sharingguidelines for options on how to legitimately share published articles. -

Ba3(P1−Xmnxo4)2 : Blue/Green Inorganic Materials Based on Tetrahedral Mn(V)
Bull. Mater. Sci., Vol. 34, No. 6, October 2011, pp. 1257–1262. c Indian Academy of Sciences. Ba3(P1−xMnxO4)2 : Blue/green inorganic materials based on tetrahedral Mn(V) SOURAV LAHA, ROHIT SHARMA, S V BHAT†, MLPREDDY‡, J GOPALAKRISHNAN∗ and S NATARAJAN Solid State and Structural Chemistry Unit, †Department of Physics, Indian Institute of Science, Bangalore 560 012, India ‡Chemical Science and Technology Division, National Institute for Interdisciplinary Science and Technology (NIIST), Thiruvananthapuram 695 019, India MS received 11 May 2011 ) 3− 2 Abstract. We describe a blue/green inorganic material, Ba3(P1−xMnxO4 2 (I) based on tetrahedral MnO4 :3d chromophore. The solid solutions (I) which are sky-blue and turquoise-blue for x ≤ 0·25 and dark green for x ≥ 0·50, 3− are readily synthesized in air from commonly available starting materials, stabilizing the MnO4 chromophore in an isostructural phosphate host. We suggest that the covalency/ionicity of P–O/Mn–O bonds in the solid solutions tunes the crystal field strength around Mn(V) such that a blue colour results for materials with small values of x. The material could serve as a nontoxic blue/green inorganic pigment. Keywords. Blue/green inorganic material; barium phosphate/manganate(V); tetrahedral manganate(V); blue/green chromophore; ligand field tuning of colour. 1. Introduction field transitions within an unusual five coordinated trigo- nal bipyramidal Mn(III). A turquoise-blue solid based on Inorganic solids displaying bright colours are important Li1·33Ti1·66O4 spinel oxide wherein the colour arises from as pigment materials which find a wide range of appli- an intervalence charge-transfer between Ti3+ and Ti4+ has cations in paints, inks, plastics, rubbers, ceramics, ena- also been described recently (Fernández-Osorio et al 2011). -
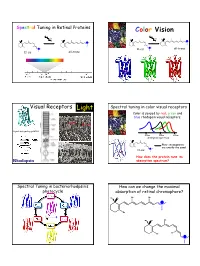
Color Vision Light
Spectral Tuning in Retinal Proteins Color Vision hν hν N N H H H H N all-trans N 11-cis 11-cis all-trans Visual Receptors Light Spectral tuning in color visual receptors Color is sensed by red, green and blue rhodopsin visual receptors. Rod Cone G-protein signaling pathway 400nm 500nm 600nm absorption spectrum Their chromophores H are exactly the same! 11-cis N How does the protein tune its Rhodopsin absorption spectrum? Spectral Tuning in bacteriorhodpsin’s How can we change the maximal photocycle absorption of retinal chromophore? Me Me Me Me N H Me Me Me Me N H Me Me Me Me Me Me Me H N 1 Excitation energy determines the Electronic Absorption maximal absorption Me Me Me Me π π* N - H S1 Me Absorption of light in the UV-VIS region of the S0 spectrum is due to Response excitation of electrons to higher energy levels. Me Me Me 13 7911 15 N H Me Me π-π* excitation in polyenes π-π* excitation in polyenes π∗ π∗ π∗ π∗ ∆E π∗ E photon E π π π π π Ground state (S0) Excited state (S1) ∆E (excitation energy, band gap) = hν = hc/λ blue-shift red-shift π-π* excitation in polyenes Tuning the length of the conjugated backbone β-carotene O O Vitamin A2 (retinal II) Vitamin A1 (retinal I) Longer wavelength Short wavelength 2 Retinal I Retinal II OPSIN SHIFT: how protein tunes the absorption maximum of its chromophore. Maximal absorption of protonated retinal Schiff base in: Water/methanol solution: 440 nm bR: 568 nm rod Rh: 500 nm red receptor: 560 nm green receptor: 530 nm blue receptor: 426 nm Salmon: different retinals in different stages of life Electrostatics and opsin shift Electrostatics and opsin shift S S positive charge 2 positive charge 2 S2 S2 Me Me Me Me Me Me S1 S1 S1 S1 + + + + N N H H Me O S0 Me O S0 Me O Me O C C Asp (Glu) Asp (Glu) S0 S0 no protein in protein no protein in protein counterion counterion • The counterion stabilizes the positive charge of the chromophore. -

Effect of Indigo Dye Effluent on the Growth, Biomass Production and Phenotypic Plasticity of Scenedesmus Quadricauda (Chlorococcales)
Anais da Academia Brasileira de Ciências (2014) 86(1): 419-428 (Annals of the Brazilian Academy of Sciences) Printed version ISSN 0001-3765 / Online version ISSN 1678-2690 http://dx.doi.org/10.1590/0001-3765201420130225 www.scielo.br/aabc Effect of indigo dye effluent on the growth, biomass production and phenotypic plasticity of Scenedesmus quadricauda (Chlorococcales) MATHIAS A. CHIA1 and RILWAN I. MUSA2 1Laboratório de Cianobactérias, Escola Superior de Agricultura Luiz de Queiroz, Universidade de São Paulo, Av. Pádua Dias, 11, 13418-900 Piracicaba, SP, Brasil 2Department of Biological Sciences, Ahmadu Bello University, Zaria, Postal Code 810001, Nigeria Manuscript received on June 26, 2013; accepted for publication on October 14, 2013 ABSTRACT The effect of indigo dye effluent on the freshwater microalga Scenedesmus quadricauda ABU12 was investigated under controlled laboratory conditions. The microalga was exposed to different concentrations of the effluent obtained by diluting the dye effluent from 100 to 175 times in bold basal medium (BBM). The growth rate of the microalga decreased as indigo dye effluent concentration increased (p <0.05). The EC50 was found to be 166 dilution factor of the effluent. Chlorophyll a, cell density and dry weight production as biomarkers were negatively affected by high indigo dye effluent concentration, their levels were higher at low effluent concentrations (p <0.05). Changes in coenobia size significantly correlated with the dye effluent concentration. A shift from large to small coenobia with increasing indigo dye effluent concentration was obtained. We conclude that even at low concentrations; effluents from textile industrial processes that use indigo dye are capable of significantly reducing the growth and biomass production, in addition to altering the morphological characteristics of the freshwater microalga S. -

(12) United States Patent (10) Patent No.: US 8,546,502 B2 Shimanaka Et Al
USOO8546502B2 (12) United States Patent (10) Patent No.: US 8,546,502 B2 Shimanaka et al. (45) Date of Patent: Oct. 1, 2013 (54) METHOD FOR PRODUCING DYE POLYMER, JP 2000-500516 A 1, 2000 DYE POLYMER AND USE OF THE SAME JP 2000-514479. A 10, 2000 JP 2000-515181 A 11, 2000 JP 2005-345512 A 12/2005 (75) Inventors: Hiroyuki Shimanaka, Chuo-ku (JP); JP 2005-352053 A 12/2005 Toshiyuki Hitotsuyanagi, Chuo-ku (JP); JP 2006-16488 * 1, 2006 Yoshikazu Murakami, Chuo-ku (JP); JP 2006-016488 A 1, 2006 JP 2006-167674 * 6, 2006 Atsushi Goto, Uji (JP); Yoshinobu JP 2006-0167674. A 6, 2006 Tsujii, Uji (JP); Takeshi Fukuda, Uji JP 2007-277533 A 10/2007 (JP) WO WO97, 18247 A1 5, 1997 WO WO98/O1478 A1 1, 1998 (73) Assignees: Dainichiseika Color & Chemicals Mfg. WO WO 98.01480 A1 1, 1998 Co., Ltd., Chuo-ku, Tokyo (JP); Kyoto WO WO99,05099 A1 2, 1999 University, Kyoto-shi, Kyoto (JP) OTHER PUBLICATIONS (*) Notice: Subject to any disclaimer, the term of this Shimizu Itaru, JP2006016488 (Jan. 2006), English Translation.* patent is extended or adjusted under 35 Shimizu Itaru et al., JP2006 167674 (Jun. 2006), English Transla U.S.C. 154(b) by 0 days. tion. Hawker, C., et al., New Polymer Synthesis by Nitroxide Mediated (21) Appl. No.: 12/737,239 Living Radical Polymerizations, Chemical Review, vol. 101, No. 12, 2001, pp. 3661-3688. (22) PCT Filed: Jun. 26, 2009 Kamigaito, M., et al., Metal-Catalyzed Living Radical Polymeriza tion, Chemical Review, vol. 101, No. -

Indigo: Sources, Processes and Possibilities for Bioregional Blue
Indigo: Sources, processes and possibilities for bioregional blue Nicholas Wenner and Matthew Forkin June 2017 Photo by Kalie Cassel-Feiss by Kalie Photo Table of Contents Introduction . .3 Indigo . .4 The Indigo Process . 11 Conclusions . 15 Photo by Paige Green Green by Paige Photo Indigo Overview 2 Introduction his report was completed with funding generously provided by the Jena and Michael King TFoundation as part of Fibershed’s True Blue project . It is one project of many that support Fibershed’s larger mission: “Fibershed develops regional and regenerative fiber systems on behalf of independent working producers, by expanding opportunities to implement carbon farming, forming catalytic foundations to rebuild regional manufacturing, and through connecting end-users to farms and ranches through public education.” In this report we present the various sources of blue dye and of indigo, and motivate the use of plant-based indigo in particular . We also identify the limitations of natural dyes like indigo and the need for larger cultural and systemic shifts . The ideal indigo dye production system would be a closed-loop system that moves from soil to dye to textiles and back to soil . The indigo process has three basic steps: planting, harvesting, and dye extraction . In this document, we provide an overview of each, and detailed explorations are given in two separate documents that will be available through Fibershed by late-summer 2017 . This report is based on a literature review of academic research, natural dye books, online content, and personal interviews . It benefited greatly from conversations with (and the generosity of) many skilled artisans and natural dyers, including Rowland Ricketts, Jane Palmer, and Kori Hargreaves . -

Red, Blue and Purple Dyes
Purple, Blue and Red Dyes We have discussed the vibrant colors of flowers, the somber colors of ants, the happy colors of leaves throughout their lifespan, the iridescent colors of butterflies, beetles and birds, the attractive and functional colors of human eyes, skin and hair, the warm colors of candlelight, the inherited colors of Mendel’s peas, the informative colors of stained chromosomes and stained germs, the luminescent colors of fireflies and dragonfish, and the abiotic colors of rainbows, the galaxies, the sun and the sky. The natural world is a wonderful world of color! The infinite number of colors in the solar spectrum was divided into seven colors by Isaac Newton—perhaps for theological reasons. While there is no scientific reason to divide the spectral colors into seven colors, there is a natural reason to divide the spectral colors into three primary colors. Thomas Young (1802), who was belittled as an “Anti-Newtonian” for speaking out about the wave nature of light, predicted that if the human eye had three photoreceptor pigments, we could perceive all the colors of the rainbow. He was right. 751 Thomas Young (1802) wrote “Since, for the reason assigned by NEWTON, it is probable that the motion of the retina is rather of a vibratory [longitudinal] than of an undulatory [transverse] nature, the frequency of the vibrations must be dependent on the constitution of this substance. Now, as it is almost impossible to conceive each sensitive point of the retina to contain an infinite number of particles, each capable of vibrating