IMFINZI, INN-Durvalumab
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fig. L COMPOSITIONS and METHODS to INHIBIT STEM CELL and PROGENITOR CELL BINDING to LYMPHOID TISSUE and for REGENERATING GERMINAL CENTERS in LYMPHATIC TISSUES
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date Χ 23 February 2012 (23.02.2012) WO 2U12/U24519ft ft A2 (51) International Patent Classification: AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, A61K 31/00 (2006.01) CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (21) International Application Number: HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, PCT/US201 1/048297 KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, (22) International Filing Date: ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, 18 August 201 1 (18.08.201 1) NO, NZ, OM, PE, PG, PH, PL, PT, QA, RO, RS, RU, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, (25) Filing Language: English TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, (26) Publication Language: English ZW. (30) Priority Data: (84) Designated States (unless otherwise indicated, for every 61/374,943 18 August 2010 (18.08.2010) US kind of regional protection available): ARIPO (BW, GH, 61/441,485 10 February 201 1 (10.02.201 1) US GM, KE, LR, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, 61/449,372 4 March 201 1 (04.03.201 1) US ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, (72) Inventor; and EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, ΓΓ, LT, LU, (71) Applicant : DEISHER, Theresa [US/US]; 1420 Fifth LV, MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, Avenue, Seattle, WA 98101 (US). -

Remarkably Similar CTLA-4 Binding Properties of Therapeutic Ipilimumab and Tremelimumab Antibodies
www.impactjournals.com/oncotarget/ Oncotarget, 2017, Vol. 8, (No. 40), pp: 67129-67139 Research Paper Remarkably similar CTLA-4 binding properties of therapeutic ipilimumab and tremelimumab antibodies Mengnan He1,2,*, Yan Chai1,*, Jianxun Qi1, Catherine W.H. Zhang3, Zhou Tong4, Yi Shi1, Jinghua Yan4, Shuguang Tan1 and George F. Gao1,2 1CAS Key Laboratory of Pathogenic Microbiology and Immunology, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China 2University of Chinese Academy of Sciences, Beijing 100049, China 3ImmuFuCell Biotechnology Co., Ltd., Beijing 100102, China 4CAS Key Laboratory of Microbial Physiological and Metabolic Engineering, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China *These authors have contributed equally to this work Correspondence to: George F. Gao, email: [email protected] Shuguang Tan, email: [email protected] Keywords: ipilimumab, CTLA-4, complex structure, tremelimumab Received: April 04, 2017 Accepted: April 21, 2017 Published: May 19, 2017 Copyright: He et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Monoclonal antibody based immune checkpoint blockade therapies have achieved clinical successes in management of malignant tumors. As the first monoclonal antibody targeting immune checkpoint molecules entered into clinics, the molecular basis of ipilimumab-based anti-CTLA-4 blockade has not yet been fully understood. In the present study, we report the complex structure of ipilimumab and CTLA-4. The complex structure showed similar contributions from VH and VL of ipilimumab in binding to CTLA-4 front β-sheet strands. -

New Biological Therapies: Introduction to the Basis of the Risk of Infection
New biological therapies: introduction to the basis of the risk of infection Mario FERNÁNDEZ RUIZ, MD, PhD Unit of Infectious Diseases Hospital Universitario “12 de Octubre”, Madrid ESCMIDInstituto de Investigación eLibraryHospital “12 de Octubre” (i+12) © by author Transparency Declaration Over the last 24 months I have received honoraria for talks on behalf of • Astellas Pharma • Gillead Sciences • Roche • Sanofi • Qiagen Infections and biologicals: a real concern? (two-hour symposium): New biological therapies: introduction to the ESCMIDbasis of the risk of infection eLibrary © by author Paul Ehrlich (1854-1915) • “side-chain” theory (1897) • receptor-ligand concept (1900) • “magic bullet” theory • foundation for specific chemotherapy (1906) • Nobel Prize in Physiology and Medicine (1908) (together with Metchnikoff) Infections and biologicals: a real concern? (two-hour symposium): New biological therapies: introduction to the ESCMIDbasis of the risk of infection eLibrary © by author 1981: B-1 antibody (tositumomab) anti-CD20 monoclonal antibody 1997: FDA approval of rituximab for the treatment of relapsed or refractory CD20-positive NHL 2001: FDA approval of imatinib for the treatment of chronic myelogenous leukemia Infections and biologicals: a real concern? (two-hour symposium): New biological therapies: introduction to the ESCMIDbasis of the risk of infection eLibrary © by author Functional classification of targeted (biological) agents • Agents targeting soluble immune effector molecules • Agents targeting cell surface receptors -

Actemra® (Tocilizumab)
Actemra® (tocilizumab) (Intravenous) Document Number: MODA-0002 Last Review Date: 10/26/2020 Date of Origin: 09/21/2010 Dates Reviewed: 12/2010, 03/2011, 05/2011, 06/2011, 09/2011, 12/2011, 03/2012, 06/2012, 09/2012, 09/2012, 11/2012, 12/2012, 03/2013, 06/2013, 09/2013, 11/2013, 12/2013, 03/2014, 06/2014, 09/2014, 12/2014, 03/2015, 05/2015, 09/2015, 12/0215, 03/2016, 06/2016, 09/2016, 12/2016, 03/2017, 05/2017, 09/2017, 12/2017, 03/2018, 06/2018, 10/2018, 10/2019, 10/2020, 11/2020 I. Length of Authorization Coverage will be provided as follows: o Castleman’s Disease: 4 months and may be renewed o Cytokine Release Syndrome: 4 doses only and may not be renewed o Immune Checkpoint Inhibitor related arthritis: 1 dose and may not be renewed o All other indications: 6 months and may be renewed. II. Dosing Limits A. Quantity Limit (max daily dose) [NDC Unit]: o Actemra 80 mg/4 mL vial: 1 vial per 14 days o Actemra 200 mg/10 mL vial: 1 vial per 14 days o Actemra 400 mg/20 mL vial: 2 vials per 14 days B. Max Units (per dose and over time) [HCPCS Unit]: Diagnosis Billable Units Interval (days) Rheumatoid Arthritis & Polyarticular Juvenile Idiopathic 800 28 Arthritis, NMOSD Systemic Juvenile Idiopathic Arthritis, Castleman’s Disease (NHL) & Acute Graft Versus Host Disease 800 14 (aGVHD) Cytokine Release Syndrome (CRS) 3200 1 course of therapy only Immune Checkpoint Inhibitor related arthritis 800 1 course of therapy only III. -

ESCMID Online Lecture Library @ by Author
The diverse monoclonal antibodies in immunology and medical oncology: relevance for infectious diseases Dra. Isabel Ruiz Camps ESCMIDHospital Online Universitari Lecture Vall d’Hebron Library @ by authorBarcelona Disclosure • Astellas • Gilead Sciences • MSD • Novartis • Pfizer ESCMID Online Lecture Library @ by author A huge topic for such a ESCMIDshort Online time Lecture Library @ by author natalizumab antiTNF antiCD20: rituximab, obinutuzumab, ofatumumab gemtuzumab (antiCD33) alemtuzumab (antiCD52) daratumumab (antiCD38) Inotuzumab (antiCD22) Brentuximab (CD30) Seculinumab (anti IL-17) Tocilizumab (antiIL6) PI3K inhibitors PARP inh: olaparib, Guselkumab (anti IL12/23) rucaparib (copanlisib, more ... Roxulitinib, tofacitinib (JAK inh) Urelumab (CD137 R) Immunotherapy: ipilimumab TK inhibitors : Imatinib, dasatinib, Belimumab (antiBAFF) (CTLA-4), tremelimumab (CTLA-4), masitinib, bosutinib, nilotinib, fostamatinib nivolimumab (PD1/PDL1), (spleen), ibrutinib (BTK), alisertib (ATK), Pembrolizumab (PD1), more...... afatinib Cabozantinib: MET, RET, VEGFR2 HER2/neu:ESCMID trastuzumab, lapatinib Online VEGFR: bevacizumabLecture, LibrarymTOR: temsirolimus sorafenib, sunitinib EGFR: cetuximab, panitumumab, MAPK inh: dabrafenib, vemurafenib erlotinib, gefitinib Selinexor (XPO1 antagonist) @ by author Trametinib (MEK inh) Index • Background • Biological therapies for immunological diseases and risk of infection • Biological therapies for cancer – Target pathways – Risk of infection • Prevention ESCMID Online Lecture Library @ by author What are -

Actemra (Tocilizumab) NON-ONCOLOGY POLICY (Intravenous) Department: PHA
Policy Title: Actemra (tocilizumab) NON-ONCOLOGY POLICY (Intravenous) Department: PHA Effective Date: 01/01/2020 Review Date: 09/25/2019, 12/18/2019, 1/22/2020, 8/3/2020 Revision Date: 09/25/2019, 12/18/2019, 1/22/2020, 8/3/2020 Purpose: To support safe, effective and appropriate use of Actemra (tocilizumab). Scope: Medicaid, Commerical, Medicare-Medicaid Plan (MMP) Policy Statement: Actemra (tocilizumab) is covered under the Medical Benefit when used within the following guidelines for non-oncology indications. Use outside of these guidelines may result in non-payment unless approved under an exception process. For oncology indications, please refer to NHPRI Oncology Policy Procedure: Coverage of Actemra (tocilizumab) will be reviewed prospectively via the prior authorization process based on criteria below. Initial Criteria: Patient has been evaluated and screened for the presence of latent TB infection prior to initiating treatment; AND Patient does not have an active infection, including clinically important localized infections; AND Must not be administered concurrently with live vaccines; AND Patient is not on concurrent treatment with another TNF-inhibitor, biologic response modifier or other non-biologic agent (i.e., apremilast, tofacitinib, baricitinib); MMP members who have previously received this medication within the past 365 days are not subject to Step Therapy Requirements Rheumatoid Arthritis Patient is 18 years or older; AND Physician has assessed baseline disease severity utilizing an objective measure/tool; AND -

On the Horizon: Immuno-Oncology (I-O) Combinations
Immuno-Oncology (I-O) Combinations • Jeffrey A. Sosman, MD • Robert H. Lurie Comprehensive Cancer Center of Northwestern University The Cancer–Immunity Cycle Daniel Chen and Ira Mellman Immunity, Volume 39, Issue 1, 2013, 1 - 10 The Cancer–Immunity Cycle Daniel Chen and Ira Mellman Immunity, Volume 39, Issue 1, 2013, 1 - 10 Stimulatory and Inhibitory Factors in the Cancer-Immunity Cycle Each step of the Cancer-Immunity Cycle requires the coordination of numerous factors, both stimulatory and inhibitory in nature. Stimulatory factors shown in green promote immunity, ... Where will Improvements come from? • Combinations: – Based on Template: anti-PD-1/PD-L1 or with anti-PD- 1/anti-CTLA-4 • Block other co-inhibitory: LAG3, TIM3, KIR, VISTA • Activate co-stimulatory: 4-1BB, OX-40, GITR, CD27, ICOS • Block inhibitory molecules- IDOi, TGFbi, CSF1Ri, anti-IL-6 or anti- IL-10 • Effect trafficking- anti-VEGF, CCL5, CXCR4i • Vaccines- TVEC- oncolytic virus, Neoantigen, other cellular • Adoptive Cellular therapy- TIL, CAR-T cells, TCR T-cells Where will Improvements come from? • Combinations: – Based on Template: anti-PD-1/PD-L1 or with anti-PD- 1/anti-CTLA-4 • Signal Inhibition, BRAF directed (BRAFi+MEKi), MEKi, PI3K inhibition (PTEN effects) • Cytokines- IL-2, IFN a,b,g,, Directed cytokines (FAP-IL-2v or CEA-IL-2v) • Epigenetic modulation- gene expression and EVR expression • Microbiome modification- fecal transplants • Chemotherapy other cytotoxics • Localized Irradiation SBRT, SRS T cells in Tumors Express Multiple Immunoinhibitory Receptors -
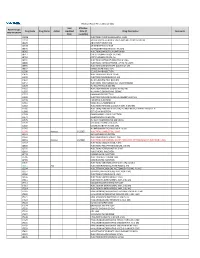
2021 Prior Authorization List Part B Appendix a (PDF)
Medicare Part B PA List Effective 2021 Last Effective Part B Drugs: Drug Code Drug Name Action Updated Date (if Drug Description Comments STEP THERAPY Date available) C9050 INJECTION, EMAPALUMAB-LZSG, 1 MG C9122 MOMETASONE FUROATE SINUS IMPLANT 10 MCG SINUVA J0129 ABATACEPT INJECTION J0178 AFLIBERCEPT INJECTION J0570 BUPRENORPHINE IMPLANT 74.2MG J0585 INJECTION,ONABOTULINUMTOXINA J0717 CERTOLIZUMAB PEGOL INJ 1MG J0718 CERTOLIZUMAB PEGOL INJ J0791 INJECTION CRIZANLIZUMAB-TMCA 5 MG J0800 INJECTION, CORTICOTROPIN, UP TO 40 UNITS J0896 INJECTION LUSPATERCEPT-AAMT 0.25 MG J0897 DENOSUMAB INJECTION J1300 ECULIZUMAB INJECTION J1428 INJECTION ETEPLIRSEN 10 MG J1429 INJECTION GOLODIRSEN 10 MG J1442 INJ FILGRASTIM EXCL BIOSIMIL J1447 INJECTION, TBO-FILGRASTIM, 1 MICROGRAM J1459 INJ IVIG PRIVIGEN 500 MG J1555 INJECTION IMMUNE GLOBULIN 100 MG J1556 INJ, IMM GLOB BIVIGAM, 500MG J1557 GAMMAPLEX INJECTION J1558 INJECTION IMMUNE GLOBULIN XEMBIFY 100 MG J1559 HIZENTRA INJECTION J1561 GAMUNEX-C/GAMMAKED J1562 INJECTION; IMMUNE GLOBULIN 10%, 5 GRAMS J1566 INJECTION, IMMUNE GLOBULIN, INTRAVENOUS, LYOPHILIZED (E.G. P J1568 OCTAGAM INJECTION J1569 GAMMAGARD LIQUID INJECTION J1572 FLEBOGAMMA INJECTION J1575 INJ IG/HYALURONIDASE 100 MG IG J1599 IVIG NON-LYOPHILIZED, NOS J1602 GOLIMUMAB FOR IV USE 1MG J1745 INJ INFLIXIMAB EXCL BIOSIMILR 10 MG J1930 Remove 1/1/2021 INJECTION, LANREOTIDE, 1 MG J2323 NATALIZUMAB INJECTION J2350 INJECTION OCRELIZUMAB 1 MG J2353 Remove 1/1/2021 INJECTION, OCTREOTIDE, DEPOT FORM FOR INTRAMUSCULAR INJECTION, 1 MG J2357 INJECTION, OMALIZUMAB, -

Looking for Therapeutic Antibodies in Next Generation Sequencing Repositories
bioRxiv preprint doi: https://doi.org/10.1101/572958; this version posted March 10, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Title: Looking for Therapeutic Antibodies in Next Generation Sequencing Repositories. Authors: Konrad Krawczyk1*, Matthew Raybould2, Aleksandr Kovaltsuk2, Charlotte M. Deane2 1 NaturalAntibody, Hamburg, Germany 2 Oxford University Department of Statistics, Oxford, UK *Correspondence to [email protected] Abstract: Recently it has become possible to query the great diversity of natural antibody repertoires using Next Generation Sequencing (NGS). These methods are capable of producing millions of sequences in a single experiment. Here we compare Clinical Stage Therapeutic antibodies to the ~1b sequences from 60 independent sequencing studies in the Observed Antibody Space Database. Of the 242 post Phase I antibodies, we find 16 with sequence identity matches of 95% or better for both heavy and light chains. There are also 54 perfect matches to therapeutic CDR-H3 regions in the NGS outputs, suggesting a nontrivial amount of convergence between naturally observed sequences and those developed artificially. This has potential implications for both the discovery of antibody therapeutics and the legal protection of commercial antibodies. Introduction Antibodies are proteins in jawed vertebrates that recognize noxious molecules (antigens) for elimination. An organism expresses millions of diverse antibodies to increase the chances that some of them will be able to bind the foreign antigen, initiating the adaptive immune response. -

NICE UPDATE for COMMISSIONERS April 2019
South, Central and West NICE UPDATE FOR COMMISSIONERS April 2019 This NICE Update for Commissioners includes: At-a-glance summary Headline update: what’s been published? Guidance and quality standards published by NICE in March 2019 What’s new for CCGs? Horizon scanning What’s coming out from NICE in the next six months? For your reference, a summary of the types of NICE guidance Reference – a guide to NICE products The next (May 2019) NICE Update for Commissioners will be issued at the beginning of June 2019. For further information about NICE guidance and its implementation contact: Tiina Korhonen, Clinical Effectiveness Lead Kathryn Markey, Clinical Effectiveness Manager Kate Forbes, Clinical Effectiveness Manager Rebecca Hodge, Clinical Effectiveness Manager Gill Barlow, Clinical Effectiveness Manager Katie Newens, Clinical Effectiveness Researcher Rachel Finch, Clinical Effectiveness Administrator [email protected] 1 | p a g e At-a-glance summary The table below shows ALL NICE guidance published in April2019. Those likely to have significant impact for CCG commissioners are discussed further in the ‘What’s new for Clinical Commissioning Groups’ section (link to relevant section provided within guidance reference). Guidance type and Title Commissioner(s) Main providers(s) Impact for CCG commissioners (financial /public reference interest/quality of care) Technology Appraisal – Daratumumab with NHS England Secondary care - TA573 bortezomib and acute and dexamethasone for Tertiary care previously treated multiple myeloma Technology Appraisal – Certolizumab pegol for CCGs Primary care, NICE does not expect this guidance to have a TA574 treating moderate to severe secondary care - significant impact on resources; that is, it will be plaque psoriasis acute and tertiary less than £5 million per year in England (or care £9,100 per 100,000 population) because the technology is an option alongside current standard treatment options and is available at a similar price. -

Tables-Of-Phase-3-Mabs.Pdf
Table 3. Monoclonal antibodies in Phase 2/3 or 3 clinical studies for cancer indications Most advanced Primary sponsoring company INN or code name Molecular format Target(s) phase Phase 3 indications Therapeutic area Janssen Research & Development, LLC JNJ-56022473 Humanized mAb CD123 Phase 2/3 Acute myeloid leukemia Cancer Murine IgG1, Actinium Pharmaceuticals Iomab-B radiolabeled CD45 Phase 3 Acute myeloid leukemia Cancer Humanized IgG1, Seattle Genetics Vadastuximab talirine ADC CD33 Phase 3 Acute myeloid leukemia Cancer TG Therapeutics Ublituximab Chimeric IgG1 CD20 Phase 3 Chronic lymphocytic leukemia Cancer Xencor XMAB-5574, MOR208 Humanized IgG1 CD19 Phase 2/3 Diffuse large B-cell lymphoma Cancer Moxetumomab Murine IgG1 dsFv, AstraZeneca/MedImmune LLC pasudotox immunotoxin CD22 Phase 3 Hairy cell leukemia Cancer Humanized scFv, Viventia Bio Oportuzumab monatox immunotoxin EpCAM Phase 3 Bladder cancer Cancer scFv-targeted liposome containing Merrimack Pharmaceuticals MM-302 doxorubicin HER2 Phase 2/3 Breast cancer Cancer MacroGenics Margetuximab Chimeric IgG1 HER2 Phase 3 Breast cancer Cancer Gastric cancer or gastroesophageal junction Gilead Sciences GS-5745 Humanized IgG4 MMP9 Phase 3 adenocarcinoma; ulcerative colitis (Phase 2/3) Cancer; Immune-mediated disorders Depatuxizumab Humanized IgG1, AbbVie mafodotin ADC EGFR Phase 2/3 Glioblastoma Cancer AstraZeneca/MedImmune LLC Tremelimumab Human IgG2 CTLA4 Phase 3 NSCLC, head & neck cancer, bladder cancer Cancer NSCLC, head & neck cancer, bladder cancer, breast AstraZeneca/MedImmune -

The Two Tontti Tudiul Lui Hi Ha Unit
THETWO TONTTI USTUDIUL 20170267753A1 LUI HI HA UNIT ( 19) United States (12 ) Patent Application Publication (10 ) Pub. No. : US 2017 /0267753 A1 Ehrenpreis (43 ) Pub . Date : Sep . 21 , 2017 ( 54 ) COMBINATION THERAPY FOR (52 ) U .S . CI. CO - ADMINISTRATION OF MONOCLONAL CPC .. .. CO7K 16 / 241 ( 2013 .01 ) ; A61K 39 / 3955 ANTIBODIES ( 2013 .01 ) ; A61K 31 /4706 ( 2013 .01 ) ; A61K 31 / 165 ( 2013 .01 ) ; CO7K 2317 /21 (2013 . 01 ) ; (71 ) Applicant: Eli D Ehrenpreis , Skokie , IL (US ) CO7K 2317/ 24 ( 2013. 01 ) ; A61K 2039/ 505 ( 2013 .01 ) (72 ) Inventor : Eli D Ehrenpreis, Skokie , IL (US ) (57 ) ABSTRACT Disclosed are methods for enhancing the efficacy of mono (21 ) Appl. No. : 15 /605 ,212 clonal antibody therapy , which entails co - administering a therapeutic monoclonal antibody , or a functional fragment (22 ) Filed : May 25 , 2017 thereof, and an effective amount of colchicine or hydroxy chloroquine , or a combination thereof, to a patient in need Related U . S . Application Data thereof . Also disclosed are methods of prolonging or increasing the time a monoclonal antibody remains in the (63 ) Continuation - in - part of application No . 14 / 947 , 193 , circulation of a patient, which entails co - administering a filed on Nov. 20 , 2015 . therapeutic monoclonal antibody , or a functional fragment ( 60 ) Provisional application No . 62/ 082, 682 , filed on Nov . of the monoclonal antibody , and an effective amount of 21 , 2014 . colchicine or hydroxychloroquine , or a combination thereof, to a patient in need thereof, wherein the time themonoclonal antibody remains in the circulation ( e . g . , blood serum ) of the Publication Classification patient is increased relative to the same regimen of admin (51 ) Int .