An Overview of Bioinformatics Tools for DNA Meta-Barcoding Analysis of Microbial Communities of Bioaerosols: Digest for Microbiologists
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Partitioning the Colonization and Extinction Components of Beta Diversity: Spatiotemporal Species Turnover Across Disturbance Gradients
bioRxiv preprint doi: https://doi.org/10.1101/2020.02.24.956631; this version posted February 25, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Research article Partitioning the colonization and extinction components of beta diversity: Spatiotemporal species turnover across disturbance gradients Shinichi TATSUMI1,2,*, Joachim STRENGBOM3, Mihails ČUGUNOVS4, Jari KOUKI4 1 Department of Biological Sciences, University of Toronto Scarborough, Toronto, ON, Canada 2 Hokkaido Research Center, Forestry and Forest Products Research Institute, Hokkaido, Japan 3 Department of Ecology, Swedish University of Agricultural Sciences, Uppsala, Sweden 4 School of Forest Sciences, University of Eastern Finland, Joensuu, Finland * Corresponding author Shinichi Tatsumi ([email protected], +1-416-208-5130) Department of Biological Sciences, University of Toronto Scarborough, 1265 Military Trail, Toronto, ON, M1C 1A4, Canada 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.02.24.956631; this version posted February 25, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. 1 ABSTRACT 2 Changes in species diversity often result from species losses and gains. The dynamic nature of 3 beta diversity (i.e., spatial variation in species composition) that derives from such temporal 4 species turnover, however, has been largely overlooked. -
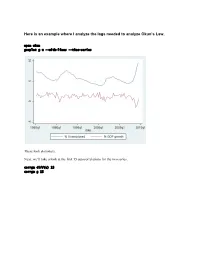
Here Is an Example Where I Analyze the Lags Needed to Analyze Okun's
Here is an example where I analyze the lags needed to analyze Okun’s Law. open okun gnuplot g u --with-lines --time-series These look stationary. Next, we’ll take a look at the first 15 autocorrelations for the two series. corrgm diff(u) 15 corrgm g 15 These are autocorrelated and so are these. I would expect that a simple regression will not be sufficient to model this relationship. Let’s try anyway. Run a simple regression and look at the residuals ols diff(u) const g series ehat=$uhat gnuplot ehat --with-lines --time-series These look positively autocorrelated to me. Notice how the residuals fall below zero for a while, then rise above for a while, and repeat this pattern. Take a look at the correlogram. The first autocorrelation is significant. More lags of D.u or g will probably fix this. LM test of residuals smpl 1985.2 2009.3 ols diff(u) const g modeltab add modtest 1 --autocorr --quiet modtest 2 --autocorr --quiet modtest 3 --autocorr --quiet modtest 4 --autocorr --quiet Breusch-Godfrey test for first-order autocorrelation Alternative statistic: TR^2 = 6.576105, with p-value = P(Chi-square(1) > 6.5761) = 0.0103 Breusch-Godfrey test for autocorrelation up to order 2 Alternative statistic: TR^2 = 7.922218, with p-value = P(Chi-square(2) > 7.92222) = 0.019 Breusch-Godfrey test for autocorrelation up to order 3 Alternative statistic: TR^2 = 11.200978, with p-value = P(Chi-square(3) > 11.201) = 0.0107 Breusch-Godfrey test for autocorrelation up to order 4 Alternative statistic: TR^2 = 11.220956, with p-value = P(Chi-square(4) > 11.221) = 0.0242 Yep, that’s not too good. -

Gamma Diversity: Derived from and a Determinant of Alpha Diversity and Beta Diversity
Acta Zoologica Mexicana (n.s.) 90: 27-76 (2003) GAMMA DIVERSITY: DERIVED FROM AND A DETERMINANT OF ALPHA DIVERSITY AND BETA DIVERSITY. AN ANALYSIS OF THREE TROPICAL LANDSCAPES Lucrecia ARELLANO y Gonzalo HALFFTER Instituto de Ecología, A.C. Departamento de Ecología y Comportamiento Animal Apartado Postal 63, 91000 Xalapa, Veracruz, MÉXICO E-mail: [email protected] [email protected] RESUMEN Utilizando tres grupos taxonómicos en este trabajo examinamos como las diversidades alfa y beta influyen en la riqueza de especies de un paisaje (diversidad gamma), así como el fenómeno recíproco. Es decir, como la riqueza en especies de un paisaje (un fenómeno histórico-biogeográfico) contribuye a determinar los valores de la diversidad alfa por sitio, por comunidad, la riqueza acumulada de especies por comunidad y la intensidad del recambio entre comunidades. Los grupos utilizados son dos subfamilias de Scarabaeoidea: Scarabaeinae y Geotrupinae, y la familia Silphidae. En todos los análisis los tres grupos taxonómicos son manejados como un grupo indicador: los escarabajos copronecrófagos. De una manera lateral se incluye información sobre la subfamilia Aphodiinae (Scarabaeoidea), escarabajos coprófagos no incorporados al manejo del grupo indicador. Los paisajes estudiados son tres (tropical, de transición y de montaña), situados en un gradiente altitudinal en la parte central del estado de Veracruz. Partimos de las premisas siguientes. La diversidad alfa de un grupo indicador refleja el número de especies que utiliza un mismo ambiente o recurso en un lugar o comunidad. La diversidad beta espacial se relaciona con la respuesta de los organismos a la heterogeneidad del espacio. La diversidad gamma depende fundamentalmente de los procesos histórico-geográficos que actúan a nivel de mesoescala y está también condicionada por las diversidades alfa y beta. -

Patterns of Alpha, Beta and Gamma Diversity of the Herpetofauna in Mexico’S Pacific Lowlands and Adjacent Interior Valleys A
Animal Biodiversity and Conservation 30.2 (2007) 169 Patterns of alpha, beta and gamma diversity of the herpetofauna in Mexico’s Pacific lowlands and adjacent interior valleys A. García, H. Solano–Rodríguez & O. Flores–Villela García, A., Solano–Rodríguez, H. & Flores–Villela, O., 2007. Patterns of alpha, beta and gamma diversity of the herpetofauna in Mexico's Pacific lowlands and adjacent interior valleys. Animal Biodiversity and Conservation, 30.2: 169–177. Abstract Patterns of alpha, beta and gamma diversity of the herpetofauna in Mexico’s Pacific lowlands and adjacent interior valleys.— The latitudinal distribution patterns of alpha, beta and gamma diversity of reptiles, amphibians and herpetofauna were analyzed using individual binary models of potential distribution for 301 species predicted by ecological modelling for a grid of 9,932 quadrants of ~25 km2 each. We arranged quadrants in 312 latitudinal bands in which alpha, beta and gamma values were determined. Latitudinal trends of all scales of diversity were similar in all groups. Alpha and gamma responded inversely to latitude whereas beta showed a high latitudinal fluctuation due to the high number of endemic species. Alpha and gamma showed a strong correlation in all groups. Beta diversity is an important component of the herpetofauna distribution patterns as a continuous source of species diversity throughout the region. Key words: Latitudinal distribution pattern, Diversity scales, Herpetofauna, Western Mexico. Resumen Patrones de diversidad alfa, beta y gama de la herpetofauna de las tierras bajas y valles adyacentes del Pacífico de México.— Se analizaron los patrones de distribución latitudinales de la diversidad alfa, beta y gama de los reptiles, anfibios y herpetofauna utilizando modelos binarios individuales de distribución potencial de 301 especies predichas mediante un modelo ecológico para una cuadrícula de 9.932 cuadrantes de aproximadamente 25 km2 cada uno. -

Drivers of Beta Diversity in Modern and Ancient Reef-Associated Soft-Bottom Environments
Drivers of beta diversity in modern and ancient reef-associated soft-bottom environments Vanessa Julie Roden1, Martin Zuschin2, Alexander Nützel3,4,5, Imelda M. Hausmann3,4 and Wolfgang Kiessling1 1 GeoZentrum Nordbayern, Section Paleobiology, Friedrich-Alexander University Erlangen-Nürnberg, Erlangen, Germany 2 Department of Palaeontology, University of Vienna, Vienna, Austria 3 SNSB—Bayerische Staatssammlung für Paläontologie und Geologie, Munich, Germany 4 Department of Earth & Environmental Sciences, Ludwig-Maximilians-Universität München, Munich, Germany 5 GeoBio-Center, Ludwig-Maximilians-Universität München, Munich, Germany ABSTRACT Beta diversity, the compositional variation among communities, is often associated with environmental gradients. Other drivers of beta diversity include stochastic processes, priority effects, predation, or competitive exclusion. Temporal turnover may also explain differences in faunal composition between fossil assemblages. To assess the drivers of beta diversity in reef-associated soft-bottom environments, we investigate community patterns in a Middle to Late Triassic reef basin assemblage from the Cassian Formation in the Dolomites, Northern Italy, and compare results with a Recent reef basin assemblage from the Northern Bay of Safaga, Red Sea, Egypt. We evaluate beta diversity with regard to age, water depth, and spatial distance, and compare the results with a null model to evaluate the stochasticity of these differences. Using pairwise proportional dissimilarity, we find very high beta diversity for the Cassian Formation (0.91 ± 0.02) and slightly lower beta diversity for the Bay of Safaga (0.89 ± 0.04). Null models show that stochasticity only plays a minor role in determining faunal differences. Spatial distance is also irrelevant. Contrary to expectations, there is no tendency of beta Submitted 26 September 2019 Accepted 16 April 2020 diversity to decrease with water depth. -

Alternative Tests for Time Series Dependence Based on Autocorrelation Coefficients
Alternative Tests for Time Series Dependence Based on Autocorrelation Coefficients Richard M. Levich and Rosario C. Rizzo * Current Draft: December 1998 Abstract: When autocorrelation is small, existing statistical techniques may not be powerful enough to reject the hypothesis that a series is free of autocorrelation. We propose two new and simple statistical tests (RHO and PHI) based on the unweighted sum of autocorrelation and partial autocorrelation coefficients. We analyze a set of simulated data to show the higher power of RHO and PHI in comparison to conventional tests for autocorrelation, especially in the presence of small but persistent autocorrelation. We show an application of our tests to data on currency futures to demonstrate their practical use. Finally, we indicate how our methodology could be used for a new class of time series models (the Generalized Autoregressive, or GAR models) that take into account the presence of small but persistent autocorrelation. _______________________________________________________________ An earlier version of this paper was presented at the Symposium on Global Integration and Competition, sponsored by the Center for Japan-U.S. Business and Economic Studies, Stern School of Business, New York University, March 27-28, 1997. We thank Paul Samuelson and the other participants at that conference for useful comments. * Stern School of Business, New York University and Research Department, Bank of Italy, respectively. 1 1. Introduction Economic time series are often characterized by positive -

3 Autocorrelation
3 Autocorrelation Autocorrelation refers to the correlation of a time series with its own past and future values. Autocorrelation is also sometimes called “lagged correlation” or “serial correlation”, which refers to the correlation between members of a series of numbers arranged in time. Positive autocorrelation might be considered a specific form of “persistence”, a tendency for a system to remain in the same state from one observation to the next. For example, the likelihood of tomorrow being rainy is greater if today is rainy than if today is dry. Geophysical time series are frequently autocorrelated because of inertia or carryover processes in the physical system. For example, the slowly evolving and moving low pressure systems in the atmosphere might impart persistence to daily rainfall. Or the slow drainage of groundwater reserves might impart correlation to successive annual flows of a river. Or stored photosynthates might impart correlation to successive annual values of tree-ring indices. Autocorrelation complicates the application of statistical tests by reducing the effective sample size. Autocorrelation can also complicate the identification of significant covariance or correlation between time series (e.g., precipitation with a tree-ring series). Autocorrelation implies that a time series is predictable, probabilistically, as future values are correlated with current and past values. Three tools for assessing the autocorrelation of a time series are (1) the time series plot, (2) the lagged scatterplot, and (3) the autocorrelation function. 3.1 Time series plot Positively autocorrelated series are sometimes referred to as persistent because positive departures from the mean tend to be followed by positive depatures from the mean, and negative departures from the mean tend to be followed by negative departures (Figure 3.1). -

Statistical Tool for Soil Biology : 11. Autocorrelogram and Mantel Test
‘i Pl w f ’* Em J. 1996, (4), i Soil BioZ., 32 195-203 Statistical tool for soil biology. XI. Autocorrelogram and Mantel test Jean-Pierre Rossi Laboratoire d Ecologie des Sols Trofiicaux, ORSTOM/Université Paris 6, 32, uv. Varagnat, 93143 Bondy Cedex, France. E-mail: rossij@[email protected]. fr. Received October 23, 1996; accepted March 24, 1997. Abstract Autocorrelation analysis by Moran’s I and the Geary’s c coefficients is described and illustrated by the analysis of the spatial pattern of the tropical earthworm Clzuniodrilus ziehe (Eudrilidae). Simple and partial Mantel tests are presented and illustrated through the analysis various data sets. The interest of these methods for soil ecology is discussed. Keywords: Autocorrelation, correlogram, Mantel test, geostatistics, spatial distribution, earthworm, plant-parasitic nematode. L’outil statistique en biologie du sol. X.?. Autocorrélogramme et test de Mantel. Résumé L’analyse de l’autocorrélation par les indices I de Moran et c de Geary est décrite et illustrée à travers l’analyse de la distribution spatiale du ver de terre tropical Chuniodi-ibs zielae (Eudrilidae). Les tests simple et partiel de Mantel sont présentés et illustrés à travers l’analyse de jeux de données divers. L’intérêt de ces méthodes en écologie du sol est discuté. Mots-clés : Autocorrélation, corrélogramme, test de Mantel, géostatistiques, distribution spatiale, vers de terre, nématodes phytoparasites. INTRODUCTION the variance that appear to be related by a simple power law. The obvious interest of that method is Spatial heterogeneity is an inherent feature of that samples from various sites can be included in soil faunal communities with significant functional the analysis. -

Applied Time Series Analysis
Applied Time Series Analysis SS 2018 February 12, 2018 Dr. Marcel Dettling Institute for Data Analysis and Process Design Zurich University of Applied Sciences CH-8401 Winterthur Table of Contents 1 INTRODUCTION 1 1.1 PURPOSE 1 1.2 EXAMPLES 2 1.3 GOALS IN TIME SERIES ANALYSIS 8 2 MATHEMATICAL CONCEPTS 11 2.1 DEFINITION OF A TIME SERIES 11 2.2 STATIONARITY 11 2.3 TESTING STATIONARITY 13 3 TIME SERIES IN R 15 3.1 TIME SERIES CLASSES 15 3.2 DATES AND TIMES IN R 17 3.3 DATA IMPORT 21 4 DESCRIPTIVE ANALYSIS 23 4.1 VISUALIZATION 23 4.2 TRANSFORMATIONS 26 4.3 DECOMPOSITION 29 4.4 AUTOCORRELATION 50 4.5 PARTIAL AUTOCORRELATION 66 5 STATIONARY TIME SERIES MODELS 69 5.1 WHITE NOISE 69 5.2 ESTIMATING THE CONDITIONAL MEAN 70 5.3 AUTOREGRESSIVE MODELS 71 5.4 MOVING AVERAGE MODELS 85 5.5 ARMA(P,Q) MODELS 93 6 SARIMA AND GARCH MODELS 99 6.1 ARIMA MODELS 99 6.2 SARIMA MODELS 105 6.3 ARCH/GARCH MODELS 109 7 TIME SERIES REGRESSION 113 7.1 WHAT IS THE PROBLEM? 113 7.2 FINDING CORRELATED ERRORS 117 7.3 COCHRANE‐ORCUTT METHOD 124 7.4 GENERALIZED LEAST SQUARES 125 7.5 MISSING PREDICTOR VARIABLES 131 8 FORECASTING 137 8.1 STATIONARY TIME SERIES 138 8.2 SERIES WITH TREND AND SEASON 145 8.3 EXPONENTIAL SMOOTHING 152 9 MULTIVARIATE TIME SERIES ANALYSIS 161 9.1 PRACTICAL EXAMPLE 161 9.2 CROSS CORRELATION 165 9.3 PREWHITENING 168 9.4 TRANSFER FUNCTION MODELS 170 10 SPECTRAL ANALYSIS 175 10.1 DECOMPOSING IN THE FREQUENCY DOMAIN 175 10.2 THE SPECTRUM 179 10.3 REAL WORLD EXAMPLE 186 11 STATE SPACE MODELS 187 11.1 STATE SPACE FORMULATION 187 11.2 AR PROCESSES WITH MEASUREMENT NOISE 188 11.3 DYNAMIC LINEAR MODELS 191 ATSA 1 Introduction 1 Introduction 1.1 Purpose Time series data, i.e. -

Quantitative Risk Management in R
QUANTITATIVE RISK MANAGEMENT IN R Characteristics of volatile return series Quantitative Risk Management in R Log-returns compared with iid data ● Can financial returns be modeled as independent and identically distributed (iid)? ● Random walk model for log asset prices ● Implies that future price behavior cannot be predicted ● Instructive to compare real returns with iid data ● Real returns o!en show volatility clustering QUANTITATIVE RISK MANAGEMENT IN R Let’s practice! QUANTITATIVE RISK MANAGEMENT IN R Estimating serial correlation Quantitative Risk Management in R Sample autocorrelations ● Sample autocorrelation function (acf) measures correlation between variables separated by lag (k) ● Stationarity is implicitly assumed: ● Expected return constant over time ● Variance of return distribution always the same ● Correlation between returns k apart always the same ● Notation for sample autocorrelation: ⇢ˆ(k) Quantitative Risk Management in R The sample acf plot or correlogram > acf(ftse) Quantitative Risk Management in R The sample acf plot or correlogram > acf(abs(ftse)) QUANTITATIVE RISK MANAGEMENT IN R Let’s practice! QUANTITATIVE RISK MANAGEMENT IN R The Ljung-Box test Quantitative Risk Management in R Testing the iid hypothesis with the Ljung-Box test ● Numerical test calculated from squared sample autocorrelations up to certain lag ● Compared with chi-squared distribution with k degrees of freedom (df) ● Should also be carried out on absolute returns k ⇢ˆ(j)2 X2 = n(n + 2) n j j=1 X − Quantitative Risk Management in R Example of -

Scale Dependence of the Beta Diversity-Scale Relationship
COMMUNITY ECOLOGY 16(1): 39-47, 2015 1585-8553/$ © AKADÉMIAI KIADÓ, BUDAPEST DOI: 10.1556/168.2015.16.1.5 Scale dependence of the beta diversity-scale relationship 1 1,4P 2 3 1 Y. ZhangP , K. MaP , M. AnandP , W. Ye and B. FuP 1 State Key Laboratory of Urban and Regional Ecology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing, 100085, China 2 Global Ecological Change Laboratory, School of Environmental Sciences, University of Guelph, Guelph, Ontario, N1G 2W1, Canada 3 Key Laboratory of Vegetation Restoration and Management of Degraded Ecosystems, South China Botanical Garden, Chinese Academy of Sciences, Guangzhou, 510650, China 4 Corresponding author. Tel/Fax: 86-10-62849104, Email: [email protected] Keywords: Alpha diversity, Diversity partitioning, Gamma diversity, Power law, Scaling. Abstract: Alpha, beta, and gamma diversity are three fundamental biodiversity components in ecology, but most studies focus only on the scale issues of the alpha or gamma diversity component. The beta diversity component, which incorporates both alpha and gamma diversity components, is ideal for studying scale issues of diversity. We explore the scale dependency of beta diversity and scale relationship, both theoretically as well as by application to actual data sets. Our results showed that a power law exists for beta diversity-area (spatial grain or spatial extent) relationships, and that the parameters of the power law are dependent on the grain and extent for which the data are defined. Coarse grain size generates a steeper slope (scaling exponent z) with lower values of intercept (c), while a larger extent results in a reverse trend in both parameters. -

Diversity Partitioning in Phanerozoic Benthic Marine Communities
Diversity partitioning in Phanerozoic benthic marine communities Richard Hofmanna,1, Melanie Tietjea, and Martin Aberhana aMuseum für Naturkunde Berlin, Leibniz Institute for Evolution and Biodiversity Science, 10115 Berlin, Germany Edited by Peter J. Wagner, University of Nebraska-Lincoln, Lincoln, NE, and accepted by Editorial Board Member Neil H. Shubin November 19, 2018 (received for review August 24, 2018) Biotic interactions such as competition, predation, and niche construc- formations harbor a pool of species that were principally able to tion are fundamental drivers of biodiversity at the local scale, yet their interact at the local and regional scale and thus can be regarded as long-term effect during earth history remains controversial. To test their the constituents of metacommunities in the geological record. role and explore potential limits to biodiversity, we determine within- Beta diversity among collections from the same formation is habitat (alpha), between-habitat (beta), and overall (gamma) diversity expected for two reasons, even though many benthic marine in- of benthic marine invertebrates for Phanerozoic geological formations. vertebrates have planktonic larval stages and thus high dispersal We show that an increase in gamma diversity is consistently generated capabilities. First, the distribution of species within a meta- by an increase in alpha diversity throughout the Phanerozoic. Beta community is predicted to be patchy (17). Even if the habitats diversity drives gamma diversity only at early stages of diversifica- represented by a formation were homogeneous, local differences tion but remains stationary once a certain gamma level is reached. This mode is prevalent during early- to mid-Paleozoic periods, in species distributions would result in compositional differences whereas coupling of beta and gamma diversity becomes increas- among sites.