Formation from Chlorophenols 82
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

First-Principles Simulations of the Interaction of Metal-Organic Molecules with a Surface and As Building Blocks for Nanodevices
Universite´ de Strasbourg IPCMS: CNRS - UMR 7504 These` de Doctorat FIRST-PRINCIPLES SIMULATIONS OF THE INTERACTION OF METAL-ORGANIC MOLECULES WITH A SURFACE AND AS BUILDING BLOCKS FOR NANODEVICES par Burak Ozdamar¨ pr´esent´eeen vue d'obtenir le grade de Docteur de l'Universit´ede Strasbourg Sp´ecialit´e:Physique de la mati`erecondens´eeet des mat´eriaux Soutenue publiquement le 28/10/2016 `al'IPCMS devant le jury compos´ede: Prof. Dr. Roberto Marquardt UdS-LCQ Pr´esident Prof. Dr. J¨urg Hutter UZH-CMSZH Rapporteur Prof. Dr. Andres Saul AMU-CINaM Rapporteur Prof. Dr. Fabrizio Cleri UL1-IEMN Rapporteur Dr. S´ebastien Le Roux UdS-IPCMS Examinateur Prof. Dr. Mauro Boero UdS-IPCMS Directeur de th`ese Tired of lying in the sunshine staying home to watch the rain You are young and life is long and there is time to kill today And then one day you find ten years have got behind you No one told you when to run, you missed the starting gun. Roger Waters iii Abstract UNIVERSITE´ DE STRASBOURG Institut de Physique et Chimie des Mat´eriauxde Strasbourg Doctor of Philosophy FIRST-PRINCIPLES SIMULATIONS OF THE INTERACTION OF METAL-ORGANIC MOLECULES WITH A SURFACE AND AS BUILDING BLOCKS FOR NANODEVICES by Burak Ozdamar¨ The purpose of this study is to investigate the interaction of organometallic com- plexes with transition metals. This topic in question has a broad array of applica- tions in a number of domain; realization of nanojunctions for molecular nanoelec- tronics, biological imaging and nanocatalysis. Within this general framework, this PhD project aims to model the fundamental interactions of molecular building blocks at the atomic level in order to understand their role in the assembly and functionalization of nanostructures. -

Metalation Studies on Titanocene Dithiolates
inorganics Article Metalation Studies on Titanocene Dithiolates Tilmann G. Kießling and Karlheinz Sünkel * ID Department of Chemistry, Ludwig-Maximilians University Munich, 81377 Munich, Germany; [email protected] * Correspondence: [email protected]; Tel.: +49-89-2180-77773 Received: 18 July 2018; Accepted: 22 August 2018; Published: 24 August 2018 Abstract: Titanocene bis-arylthiolates [(C5H4X)(C5H4Y)Ti(SC6H4R)2] (X,Y = H, Cl; R = H, Me) can be prepared from the corresponding titanocene dichlorides by reacting with the thiols in the presence of DABCO as a base. They react with n-butyl lithium to give unstable Ti(III) radical anions. While the unsubstituted thiolates (X = Y = R = H) react with lithium Di-isopropylamide by decomposing to dimeric fulvalene-bridged and thiolate-bridged Ti(III) compounds, the ring-chlorinated compounds can be deprotonated with LDA and give appropriate electrophiles di-substituted and tri-substituted titanocene dithiolates. Keywords: titanocene thiolates; metalation; chlorocyclopentadienyl ligands 1. Introduction Titanocene compounds, i.e., compounds of the type “Cp2TiXn” where “Cp” stands for a substituted or unsubstituted cyclopentadienyl ligand and “X” for any anionic or neutral ligand and n = 0–2 are arguably the second-most studied metallocenes after ferrocene. Soon after the first synthesis of (C5H5)2TiCl2 in 1954 [1], its potential for acting as a polymerization catalyst when combined with certain aluminum compounds was discovered [2]. Further studies showed that a judicious choice of substituents on the cyclopentadienyl ring had a great influence on the stereochemistry of the produced polymers, which gave rise to an enormous number of publications and also many review articles [3–8]. -

CURRICULUM VITAE WILLIAM E. GEIGER, JR. Professor Of
CURRICULUM VITAE WILLIAM E. GEIGER, JR. Professor of Chemistry University of Vermont, Burlington VT 05405 802-656-0268; EMAIL [email protected] FAX 802-656-8705 Education B.S. Canisius College 1965 Chemistry Ph.D. Cornell University 1969 Analytical Chemistry Positions Research Associate, Univ. of California at Riverside 1968-69 Postdoctoral Research Associate, Northwestern Univ. 1969-70 Assistant Professor, Southern Illinois Univ. 1970-74 Assistant Professor, Univ. of Vermont 1974-77 Associate Professor, Univ. of Vermont 1977-82 Professor, Univ. of Vermont 1982-Present Pomeroy Professor, Univ. of Vermont 1997-Present Chair, Department of Chemistry, University of Vermont 1998-2001 Honors University of Vermont Scholar 1984 James Crowdle Award, Canisius College 1995 Dean’s Lecturer, UVM 2009 Vermont Academy of Science and Engineering 2009 William Geiger Research Laboratory (Free State University) 2011 Professional Organizations Member, American Chemical Society (ACS) Division of Analytical Chemistry Division of Inorganic Chemistry Society for Electroanalytical Chemistry (SEAC) Vermont Academy of Science and Engineering Selected Professional Activities Professeur Associe, Univ. of Bordeaux 1986 Editor, Newsletter of SEAC 1985-88 Editorial Advisory Board, Organometallics 1989-91 Board of Directors, SEAC 1992-95 Graduiertenkolleg Lecturer, Univ. of Freiburg 1996 Editorial Board, Chemtracts 1997-98 Leverhulme Fellow, University of Bristol 1998 Erskine Fellow, University of Canterbury 2000 Advisory Board, Center for Molecular Electrocatalysis 2009-present INVITED LECTURES AND COLLOQUIA Canisius College 10/70 Univ. Maryland at Baltimore 9/71 Franklin and Marshall College 9/71 Univ. of Missouri at St. Louis 2/73 Brown University 10/75 Univ. of Massachusetts at Amherst 12/75 Univ. of Massachusetts at Boston 2/76 SUNY at Buffalo 11/76 SUNY at Plattsburgh 1/77 Providence College 2/78 Wesleyan University 2/78 St. -

AROMATIC HYDROCARBONS: Part I
AROMATIC HYDROCARBONS: Part I DR SUNIL K. SINGH Assistant Professor in Chemistry SK SINGH, KMC Concept of Aromaticity Aromatic compounds apparently contain alternate double and single bonds in a cyclic structure and resemble benzene in chemical behaviour. They undergo substitution rather than addition reactions. This characteristic behaviour is known as aromaticity. Addition reaction: Substitution reaction: Br Br 2 H E CCl + 4 Br E + H+ Br2 No Reaction SK SINGH, KMC CCl4 Following are the main criteria of aromaticity: Chemical behaviour: electrophilic aromatic substitution. Structural: bond length equalization due to cyclic delocalization of electrons. C-C bond length in benzene id 139 pm. SK SINGH, KMC Contd……………….. Enhanced stability (large resonance energy). Heat of hydrogenation of Benzene is 49.8 Kcal/mole. Heat of hydrogenation of 1,3,5- hexatriene is 85.8 Kcal/ mole. Smaller the heat of hydrogenation Magnetic: "ring current" effects. more is the stability. Benzene is ~36 Kcal/mole more stable than the 1,3,5- • anomalous chemical shifts in NMR. hexatriene. • large magnetic anisotropies • high diamagnetic susceptibility. SK SINGH, KMC Huckel’s Rule and aromaticity A molecule is aromatic if all the following conditions are fulfilled: It is cyclic, planar and it has continuous delocalization of p electrons (electrons in p orbitals) with or without the participation of lone pair(s)/ -ve charge/ +ve charge. Huckel’s rule: The delocalised p-electron system must contain a total of (4n+2)p electrons, where n is a whole number (i.e., n = 0,1,2,3,………). Note that the p-orbitals in which the electrons are delocalizing must be parallel to each other so that a continuous overlap of electrons is possible along the ring. -

Synthesis of the Metallocenes for the Production of Exotic High Energy Ion Beams
Synthesis of the Metallocenes for the Production of Exotic High Energy Ion Beams Ntombizonke Yvonne Kheswa A thesis is submitted in fulfilment of the requirements for the degree of Doctor of Philosophy in the Department of Physics & Astronomy, University of the Western Cape, South Africa. Supervised by: Prof. J. N. Orce, Department of Physics & Astronomy University of the Western Cape Prof. S. Titinchi, Department of Chemistry, University of the Western Cape Dr. R. Thomae Accelerator and Engineering Department iThemba LABS March 2019 https://etd.uwc.ac.za DECLARATION I declare that Synthesis of the Metallocenes for the Production of Exotic High Energy Ion Beams is my own work, that it has not been submitted for any degree or examination in any other university, and that all the sources I have used or quoted have been indicated and acknowledged by complete references. Signed: Ntombizonke Kheswa Date: 1 March 2019 i https://etd.uwc.ac.za Synthesis of the Metallocenes for the Production of Exotic High Energy Ion Beams Department of Physics and Astronomy, University of the Western Cape, Private Bag X17, 7535 Bellville, South Africa. ABSTRACT The Subatomic Physics Department of iThemba Laboratory for Accelerated Based Sciences (iThemba LABS) conducts experiments that require a variety of particle beams in order to study nuclear properties (reaction, structure, etc.) of various nuclides. These particle beams are accelerated using the K-200 Separated Sector Cyclotron (SSC) and delivered to different physics experimental vaults. Prior to acceleration, the particle beam is first ionised using an Electron Resonance Ion Source (ECRIS). The main goal of this study is the production of exotic metallic beams of 60Ni8+ and 62Ni8+ using ECRIS4, which are required for the Coulomb excitation experiments approved by the Programme Advisory Committee (PAC) at iThemba LABS. -

MITOCW | 2. the Ferrocene Lecture
MITOCW | 2. The Ferrocene Lecture [SQUEAKING] [RUSTLING] [CLICKING] JOHN DOLHUN: Welcome to the ferrocene lecture. We are going to do a demonstration at the start because I'm going to be talking about additional reactions during this lecture. And I want you to actually see one happen. So I've got Amanda Trainor, who is manning the fire extinguisher. Amanda just had her fire extinguisher testing. So she is ready to go. And Dr. Sarah Hewett will actually assist me with the bromine that we're using. Bromine and me go way back because I made bromine when I was in high school. In the parents of my-- in my parents' home-- in the basement. The whole place filled up with reddish brown gas. And back in those days, the chemistry sets had real chemicals in them. So what we're going to do is we are going to cook some bacon. And we are going to crack the glycerol. And we going to make this unsaturated compound acrolein. Then we're going to throw it into the bromine. And the bromine should add across the double bond-- an addition reaction. This is the acrid smelly stuff that you smell when you cook bacon. This is the burned bacon fat smell that you smell. It's colorless, flammable, and very poisonous. So let's go. We're going to start this. So I'm using a creme brulee torch. It's my wife's. She's probably out of her mind now looking for it. So we're cooking the bacon. -

N-Metal Compounds
31 N-METAL COMPOUNDS n the 85 years following Kekulk's brilliant proposal for the structure of ben- zene, organic chemistry underwent a tremendous expansion, and in the process a wide variety of paradigms or working hypotheses were developed about what kinds of compounds could "exist" and what kinds of reactions could occur. In many cases, acceptance of these hypotheses appeared to stifle many possible lines of investigation and caused contrary evidence to be pigeonholed as "interesting but not conclusive." As one example, the paradigm of angle strain was believed to wholly preclude substances that we know now are either stable or important reaction intermediates, such as cubane (Section 12- 1O), cyclo- propanone (Section 17-1 I), and benzyne (Sections 14-6C and 23-8). No paradigm did more to retard the development of organic chemistry than the notion that, with a "few" exceptions, compounds with bonds between carbon and transition metals (Fe, Co, Ni, Ti, and so on) are inherently un- stable. This idea was swept away in 1951 with the discovery of ferrocene, (C,H,),Fe, by P. L. Pauson. Ferrocene has unheard of properties for an organoiron compound, stable to more than 500" and able to be dissolved in, and recovered from, concentrated sulfuric acid! Pauson's work started an avalanche of research on transition metals in the general area between organic and inorganic chemistry, which has flourished ever since and has led to an improved understanding of important biochemical processes. 31-1 Metallocenes 31-1 METALLOCENES The discovery of ferrocene was one of those fortuitous accidents that was wholly unforeseeable- the kind of discovery which, over and over again, has changed the course of science. -

"Mixed-Ligand" Homocyclopentadienyl Ruthenocenes
Western Michigan University ScholarWorks at WMU Master's Theses Graduate College 12-1992 Synthesis of Symmetric and "Mixed-Ligand" Homocyclopentadienyl Ruthenocenes John Adjeiku Amanfu Follow this and additional works at: https://scholarworks.wmich.edu/masters_theses Part of the Organic Chemistry Commons Recommended Citation Amanfu, John Adjeiku, "Synthesis of Symmetric and "Mixed-Ligand" Homocyclopentadienyl Ruthenocenes" (1992). Master's Theses. 902. https://scholarworks.wmich.edu/masters_theses/902 This Masters Thesis-Open Access is brought to you for free and open access by the Graduate College at ScholarWorks at WMU. It has been accepted for inclusion in Master's Theses by an authorized administrator of ScholarWorks at WMU. For more information, please contact [email protected]. SYNTHESIS OF SYMMETRIC AND "MDCED-LIGAND" HOMOCYCLOPENTADEENYL RUTHENOCENES John Adjeiku Amanfu A Thesis Submitted to the Faculty of The Graduate College in partial fulfillment of the requirements for the Degree of Master of Arts Department of Chemistry Western Michigan University Kalamazoo, Michigan December 1992 Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. SYNTHESIS OF SYMMETRIC AND "MIXED-LIGAND HOMOCYCLOPENTADIENYL RUTHENOCENES John Adjeiku Amanfu, M.A. Western Michigan University, 1992 Muller and co-workers were the first to report the synthesis of a symmetric bicyclic analogue of ferroene, by treating the anion of bicy- cio[3.2.2]nona-2,6,8-triene (BCNT) with ferrous chloride. This compound was found to be thermally less stable than ferrocene decomposing above 50°C. We report the synthesis and characterization of two homocy- clopentadienyl ruthenocenes; namely (pentamethylcyclopentadienyl)- (bicyclo[3.2.1]octa-2,6-dienyl) ruthenium(II), {ri^-(C5Me5)Ru(BCOD)} and (cyclopentadienyl)(bicyclo[3.2.1]octa-2,6-dienyl)ruthenium(II) {^-(CsHs)- Ru(BCOD)} by treating bicyclo[3.2.1]octa-2,6-dienyl anion with the ruthenium complexes, [Cp*RuCl2]2 and [(NBD)RuCl 2 ]2 respectively. -

Transition Metal and Lanthanide Chemistry of Dipyridylpyrrolide Ligands
TRANSITION METAL AND LANTHANIDE CHEMISTRY OF DIPYRIDYLPYRROLIDE LIGANDS James McPherson A thesis presented in fulfilment of the requirements for the degree of Doctor of Philosophy School of Chemistry Faculty of Science June 2019 Thesis/Dissertation Sheet Surname/Family Name : McPherson Given Name/s : James Abbreviation for degree as give in the University calendar : PhD Faculty : Science School : Chemistry Thesis Title : Transition metal and lanthanide chemistry of dipyridylpyrrolide ligands Abstract Pincer ligands attract great current interest because they bestow exceptional stability on their metal complexes. Modification of the pincer scaffold can be used to tune the electron density at the metal ion and, therefore, the physical and chemical properties. This thesis reports studies of 2,5-di(2- pyridyl)pyrrolide (dpp–) NNN-pincer ligands and their transition and rare-earth metal complexes. Homoleptic complexes were prepared and screened for useful physical (e.g., magnetic or photophysical) or chemical (e.g., redox or reactive) properties. Predictable control of the properties through the substitution of the pyrrolide ring was a particular goal, and was achieved. The critical review of the literature on the chemistry of dpp– ligands and related pyridine-pyrrolide pincers, Chapter 1, concluded that a range of dpp– derivatives with flexible coordination modes was accessible, but with underdeveloped coordination chemistry. – Chapter 2 provides a theoretical quantification (B3LYP-GD3BJ/6-31G** calculations) of the internal stain (∆EL(strain)) that builds when a dpp or related pyridine-pyrrolide pincer ligand binds a metal ion. A relationship was discovered correlating ∆EL(strain) and the change in interatomic separation of the two terminal donor atoms when bound (ρL(coord)) and metal-free (ρL(relax)). -

Reactivity of Transition Metal Organometallics L. J. Farrugia Msc
L.J. Farrugia : MSc Core2 Course C5 - Reactivity of Transition Metal Organometallics Reactivity of Transition Metal Organometallics L. J. Farrugia MSc Core Course C5 Text books : Inorganic Chemistry - Housecroft & Sharpe Ch 23 - the very basics Inorganic Chemistry - Shriver & Atkins Ch 16 - the very basics This course assumes familiarity with the Level-2 and Level-3 courses on Organometallic Chemistry, and this is covered in the above texts Organometallics - Elschenbroich & Salzer (library) - much more useful Topic 1 - Introduction to Cyclopentadienyl Compounds First part of course covers the role of the ligand cyclopentadienyl (Cp) and its derivatives. Cp is one of the most important ligands in organometallics after CO. A considerable percentage of organometallic compounds contain this ligand - it is also a good ligand for main group metals and the f-block metals (lanthanides & actinides). H H H Fe CO H OC CO Cyclopentadiene Cyclopentadiene complex (4 e donor ligand) very rare as a ligand Na / -H2 H - Na+ H acidic H atom planar aromatic (6-pi electron) dienyl anion - The anion C 5H5 is a very useful synthetic reagent. It is usually treated as equivalent to occupying THREE coordination sites, so that C 5H5 ≡ 3(CO). In electron counting terms, it can be treated as either as a 6-e donor ANION or a 5- e donor NEUTRAL molecule. The latter is the recommended approach because it is simpler (do not need to worry about oxidation levels). 1.1 Bonding in cyclopentadienyl compounds Cp has 5 π electrons in the 5 out-of-plane p-orbitals on the C atoms. These 5 orbitals combine as a 1 + e 1 + e 2 under five-fold symmetry. -
A Qualitative Molecular Orbital Diagram for Ferrocene
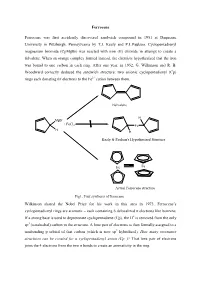
Ferrocene Ferrocene was first accidently discovered sandwich compound in 1951 at Duquesne University in Pittsburgh, Pennsylvania by T.J. Kealy and P.L.Paulson. Cyclopentadienyl magnesium bromide (CpMgBr) was reacted with iron (II) chloride in attempt to create a fulvalene. When an orange complex formed instead, the chemists hypothesized that the iron was bound to one carbon in each ring. After one year, in 1952, G. Wilkinson and R. B. Woodward correctly deduced the sandwich structure: two anionic cyclopentadienyl (Cp) rings each donating 6π electrons to the Fe2+ cation between them. Fulvalene H MgBr + FeCl2 Fe H H Kealy & Paulson's Hypothesized Structure Fe Fe Actual Ferrocene structure Fig1. First synthesis of ferrocene Wilkinson shared the Nobel Prize for his work in this area in 1973. Ferrocene’s cyclopentadienyl rings are aromatic – each containing 6 delocalized π electrons like benzene. If a strong base is used to deprotonate cyclopentadiene (Cp), the H+ is removed from the only sp3 (tetrahedral) carbon in the structure. A lone pair of electrons is then formally assigned to a nonbonding p orbital of that carbon (which is now sp2 hybridized). How many resonance structures can be created for a cyclopentadienyl anion (Cp−)? That lone pair of electrons joins the 4 electrons from the two π bonds to create an aromaticity in the ring. :B H H H : Fig 2. Aromaticity of cyclopentadienyl ring Properties of Ferrocene: [1] Ferrocene is the first sandwitch compound discovered accidently in 1951. [2] Ferrocene is a diamagnetic yellow crystalline solid in which Fe is exist as zero oxidation state. [3] The melting point of ferrocene is 174 0C and boiling point is 249 0C. -

Transition Metal Complexes with Functionalized Silyl-Substituted Cyclopentadienyl and Related Ligands: Synthesis and Reactivity
Coordination Chemistry Reviews 193–195 (1999) 447–498 www.elsevier.com/locate/ccr View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Biblioteca Digital de la Universidad de Alcalá Transition metal complexes with functionalized silyl-substituted cyclopentadienyl and related ligands: synthesis and reactivity Toma´s Cuenca, Pascual Royo * Departamento de Quı´mica Inorga´nica, Uni6ersidad de Alcala´, Campus Uni6ersitario, Edificio de Farmacia, 28871-Alcala´ de Henares, Madrid, Spain Received 7 January 1999; received in revised form 12 March 1999; accepted 19 April 1999 Contents Abstract.................................................... 448 1. Introduction ............................................... 449 2. Hydro–silyl–cyclopentadienyl metal complexes (type I) ...................... 451 2.1 Synthetic methods ......................................... 451 2.1.1 Synthesis of hydro–silyl–cyclopentadiene and related derivatives and their alkali and thallium salts ................................. 451 2.1.2 Synthesis of monocyclopentadienyl-type derivatives using diene-reagents ..... 452 2.1.3 Synthesis of dicyclopentadienyl-type derivatives using dienyl-reagents ....... 453 2.1.4 Metallation of coordinated cyclopentadienyl ligands ................. 454 2.1.5 Use of doubly silyl-bridged cyclopentadiene ...................... 455 2.2 Reactivity .............................................. 455 3. Halo–silyl–cyclopentadienyl metal complexes (type II)....................... 457 3.1 Synthetic methods ........................................