Molecular Modeling of Phase Behavior and Microstructure of Acetone-Chloroform-Methanol Binary Mixtures
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Sds – Safety Data Sheet
Effective Date: 02/05/16 Replaces Revision: 01/01/13, 02/26/09 NON-EMERGENCY TELEPHONE 24-HOUR CHEMTREC EMERGENCY TELEPHONE 610-866-4225 800-424-9300 SDS – SAFETY DATA SHEET 1. Identification Product Identifier: ACETONE / ISOPROPYL ALCOHOL BLEND Synonyms: None Chemical Formula: Not Applicable to mixtures Recommended Use of the Chemical and Restrictions On Use: Laboratory Reagent Manufacturer / Supplier: Puritan Products; 2290 Avenue A, Bethlehem, PA 18017 Phone: 610-866-4225 Emergency Phone Number: 24-Hour Chemtrec Emergency Telephone 800-424-9300 2. Hazard(s) Identification Classification of the Substance or Mixture: Flammable liquids (Category 2) Skin irritation (Category 3) Eye irritation (Category 2A) Specific target organ toxicity - single exposure (Category 3) Risk Phrases: R11: Highly flammable. R36: Irritating to eyes. R66: Repeated exposure may cause skin dryness or cracking. R67: Vapors may cause drowsiness and dizziness. Label Elements: Trade Name: ACETONE / ISOPROPYL ALCOHOL BLEND Signal Word: Danger Hazard Statements: H225: Highly flammable liquid and vapor. H316: Causes mild skin irritation. H319: Causes serious eye irritation. H336: May cause drowsiness or dizziness. ACETONE / ISOPROPYL ALCOHOL BLEND Page 1 of 6 Precautionary Statements: P210: Keep away from heat/sparks/open flames/hot surfaces. No smoking. P261: Avoid breathing dust / fume / gas / mist / vapors / spray. P305 + P351 + P338: IF IN EYES: Rinse cautiously with water for several minutes. Remove contact lenses, if present and easy to do. Continue rinsing. 3. Composition / Information on Ingredients CAS Number: Not Applicable to mixtures Molecular Weight: Not Applicable to mixtures Ingredient CAS Number EC Number Percent Hazardous Chemical Characterization Acetone 67 - 64 - 1 200-662-2 70 - 90% Yes Substance Isopropyl Alcohol 67 - 63 - 0 200-661-7 10 - 30% Yes Substance 4. -

SAFETY DATA SHEET Acetone
SAFETY DATA SHEET Acetone Revision date: 12.05.15 SDS/004/8 Page 1 of 6 1. Identification of the substance/mixture and of the company/undertaking 1.1. Product identifier Acetone EC No. 200-662-2 1.2. Relevant identified uses of the substance or mixture and uses advised against Household Solvent 1.3. Details of the supplier of the safety data sheet Thornton & Ross Ltd, Linthwaite, Huddersfield, HD7 5QH Tel: 01484 842217 Fax: 01484 847301 Email: [email protected] 1.4 Emergency telephone number: Out of normal working hours: +44 870 8510207 2. Hazards identification 2.1. Classification of the substance or mixture According to Regulation EC 1272/2008 classified as Flammable Liquid Category 2, Eye Irritant Category 2, Specific Target Organ Toxicity Single Exposure Category 3. 2.2. Label element GHS Pictogram Signal Word Hazard Class Danger Flammable Liquids, Category 2 Eye Irritation, Category 2 Specific Target Organ Toxicity-Single Exposure, Category 3 Hazard Statements Precautionary statements H225: Highly flammable liquid and vapour. P210: Keep away from heat/sparks/open flames/hot surfaces – No smoking. H319: Causes serious eye irritation P305+351+338: IF IN EYES: Rinse cautiously with H336: May cause drowsiness or dizziness water for several minutes. Remove contact lenses if present and easy to do – continue rinsing. P337+313: Get medical advice/attention. P403: Store in a well ventilated place. Supplemental Hazard Information (EU) EUH066: Repeated exposure may cause skin dryness or cracking 2.3. Other hazards N/A SAFETY DATA SHEET Acetone Revision date: 12.05.15 SDS/004/8 Page 2 of 6 3. -

Toxicological Profile for Acetone Draft for Public Comment
ACETONE 1 Toxicological Profile for Acetone Draft for Public Comment July 2021 ***DRAFT FOR PUBLIC COMMENT*** ACETONE ii DISCLAIMER Use of trade names is for identification only and does not imply endorsement by the Agency for Toxic Substances and Disease Registry, the Public Health Service, or the U.S. Department of Health and Human Services. This information is distributed solely for the purpose of pre dissemination public comment under applicable information quality guidelines. It has not been formally disseminated by the Agency for Toxic Substances and Disease Registry. It does not represent and should not be construed to represent any agency determination or policy. ***DRAFT FOR PUBLIC COMMENT*** ACETONE iii FOREWORD This toxicological profile is prepared in accordance with guidelines developed by the Agency for Toxic Substances and Disease Registry (ATSDR) and the Environmental Protection Agency (EPA). The original guidelines were published in the Federal Register on April 17, 1987. Each profile will be revised and republished as necessary. The ATSDR toxicological profile succinctly characterizes the toxicologic and adverse health effects information for these toxic substances described therein. Each peer-reviewed profile identifies and reviews the key literature that describes a substance's toxicologic properties. Other pertinent literature is also presented, but is described in less detail than the key studies. The profile is not intended to be an exhaustive document; however, more comprehensive sources of specialty information are referenced. The focus of the profiles is on health and toxicologic information; therefore, each toxicological profile begins with a relevance to public health discussion which would allow a public health professional to make a real-time determination of whether the presence of a particular substance in the environment poses a potential threat to human health. -

Annexes To: CPMP/ICH/283/95 Impurities: Guideline for Residual Solvents & CVMP/VICH/502/99 Guideline on Impurities
20 February 2013 CPMP/QWP/450/03 -Rev.1, EMEA/CVMP/511/03 -Rev.1 Committee for medicinal products for human use (CHMP) Committee for medicinal products for veterinary use (CVMP) Annexes to: CPMP/ICH/283/95 Impurities: Guideline for residual solvents & CVMP/VICH/502/99 Guideline on impurities: residual solvents Annex I: specifications for class 1 and class 2 residual solvents in active substances Annex II: residues of solvents used in the manufacture of finished products Discussion at Quality Working Party January 2003 to June 2004 Adoption by CVMP July 2004 Adoption by CHMP July 2004 Date for coming into operation January 2005 Rev. 01 Adoption by Quality Working Party 22 November 2012 Rev. 01 Adoption by CVMP 7 February 2013 Rev. 01 Adoption by CHMP 11 February 2013 Rev. 01 Date for coming into operation 1 March 2013 7 Westferry Circus ● Canary Wharf ● London E14 4HB ● United Kingdom Telephone +44 (0)20 7418 8400 Facsimile +44 (0)20 7418 8416 E -mail [email protected] Website www.ema.europa.eu An agency of the European Union © European Medicines Agency, 2013. Reproduction is authorised provided the source is acknowledged. Annexes to: CPMP/ICH/283/95 Impurities: Guideline for residual solvents & CVMP/VICH/502/99 Guideline on impurities: residual solvents Introduction The two (V)ICH residual solvents guidelines, ICH Q3C Impurities: Guideline for residual solvents (CPMP/ICH/283/95) and VICH GL18 Guideline on impurities: residual solvents in new veterinary medicinal products, active substances and excipients (CVMP/VICH/502/99), have been in operation for several years, since March 1998 and June 2001 respectively. -

Acetone - Toxfaqs™
Acetone - ToxFAQs™ What is acetone? Acetone is a (manmade) chemical that can also be found in the environment. It is a colorless liquid with a distinct smell and taste. It evaporates easily in air, is flammable, and dissolves in water. Acetone is used by humans to dissolve other substances and to produce plastics, paints and coatings, cleaning products, and personal care products. Other manmade sources of acetone are vehicle exhaust, tobacco smoke, and landfills. Acetone is also released naturally by plants, trees, insects, microbes (germs), volcanic erruptions, and forest fires, and can be found naturally in many fruits and vegetables. Low levels of acetone are produced naturally by the human body, and some health conditions can cause these levels to increase. What happens to acetone in the environment? • Most acetone in the environment exists as vapor in the air, and it can travel long distances this way. • About half of the total acetone in air is broken down by sunlight or other chemicals within 22 days. • Acetone moves from the air into water and soil by rain and snow, and moves quickly from water and soil back into the air. It does not bind to soil or build up in animals. • Acetone can enter surface water as manufacturing waste and seep into groundwater from landfills. • Acetone is broken down by microbes or chemicals in water and soil. How can I be exposed to acetone? A strong scent of acetone • Low levels of acetone are in the air, so most people are exposed and irritation in your eyes, to very small amounts through breathing, but these are rarely at nose, and throat are levels that are harmful to your health. -
Extractive Distillation of Acetone-Methanol Mixture Using Dimethyl Sulfoxide As Entrainer
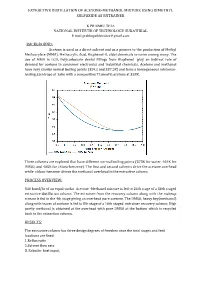
EXTRACTIVE DISTILLATION OF ACETONE-METHANOL MIXTURE USING DIMETHYL SULFOXIDE AS ENTRAINER K PRABHU TEJA NATIONAL INSTITUTE OF TECHNOLOGY SURATHKAL E mail:[email protected] BACKGROUND: Acetone is used as a direct solvent and as a pioneer to the production of Methyl Methacrylate (MMA), Methacrylic Acid, Bisphenol-A, aldol chemicals to name among many. The use of MMA in LCD, Polycarbonate dental fillings from Bisphenol play an indirect role of demand for acetone in consumer electronics and industrial chemicals. Acetone and methanol have very similar normal boiling points (329.2 and 337.5K) and form a homogeneous minimum- boiling azeotrope at 1atm with a composition77.6mol% acetone at 328K. Three solvents are explored that have different normal boiling points (373K for water, 464K for DMSO, and 405K for chloro-benzene). The rst and second solvents drive the acetone overhead while chloro-benzene drives the methanol overhead in the extractive column. fi PROCESS OVERVIEW: 540 kmol/hr of an equal-molar Acetone -Methanol mixture is fed to 24th stage of a 36th staged extractive distillation column. The entrainer from the recovery column along with the makeup stream is fed to the 4th stage giving an overhead pure acetone. The DMSO, heavy key(methanol) along with traces of acetone is fed to 8th stage of a 16th staged entrainer recovery column. High purity methanol is obtained at the overhead with pure DMSO at the bottom which is recycled back to the extraction column. RESULTS: The extractive column has three design degrees of freedom once the total stages and feed locations are fixed: 3..Reflux Reboiler ratio heat input. -

Incompatible Chemical Groups.Pdf
Incompatible Chemical Hazard Groups (and some common examples) Mineral Acids Do NOT Store with… Hydrochloric acid Hydrogen peroxide Acetone Sulfuric Acid Sodium hydroxide Methanol Phosphoric Acid Calcium hydroxide Nitric Acid (keep separate) Chloroform Acetic Acid Strong Organic Acids Do NOT Store with… Acetic Acid3, 4 Hydrogen peroxide Acetone Acetonitrile Formic Acid Sodium hydroxide Methanol Benzene Sulfuric Acid Chloroform Special 1. Organic acids are varied and may be incompatible with each other. Notes: Check MSDSs for specifics 2. Store nitric acid separately in its own secondary container. It is a strong oxidizer. 3. Store acetic acid away from oxidizing agents — especially nitric acid. 4. Acetic acid may be stored with some inorganic acids and most flammable solvents but keep in a separate secondary container. (>70% acetic acid is combustible). Weak These are typically not corrosive and not strongly reactive and can be Organic Acids stored with general liquid lab chemicals. Examples include butyric, maleic, and benzoic acids. Non-Flammable Do NOT Store with… Chlorinated Solvents Methylene chloride Acetone Hexane Chloroform Methanol Nitric Acid Trichloroethane Ethanol Hydrogen Peroxide Carbon tetrachloride Organic Solvents Do NOT Store with… Acetone Hydrogen peroxide Nitric Acid Methanol Sodium hydroxide Chromic Acid Phenol Calcium hydroxide Sulfuric Acid Xylene Trichlorfluoromethane Hydrochloric Acid Oxidizers Do NOT Store with… Nitric Acid Sodium metal Paper and oily rags Hydrogen peroxide Isopropyl Alcohol Xylene Chromic Acid Acetone Sodium nitrate Perchloric Acid Ethyl ether Bromate salts . -

Steady State Design for the Separation of Acetone-Chloroform Maximum Boiling Azeotrope Using Three Different Solvents
Korean Chem. Eng. Res., 55(4), 490-496 (2017) https://doi.org/10.9713/kcer.2017.55.4.490 PISSN 0304-128X, EISSN 2233-9558 Steady State Design for the Separation of Acetone-Chloroform Maximum Boiling Azeotrope Using Three Different Solvents Manish Pokhrel, Asante Daniel Owusu, and Jungho Cho† Department of Chemical Engineering, Kongju National University, 1223-24, Cheonan-daero, Seobuk-gu, Cheonan, Chungnam, 31080, Korea (Received 26 October 2016; Received in revised form 30 March 2017; accepted 31 March 2017) Abstract − We have designed an extractive distillation for separating maximum boiling azeotrope of acetone-chloro- form system. PRO/II 9.4 was used to simulate the overall process. The VLE data adopted from Dortmund data bank was regressed to obtain a new set of binary interaction parameters. Three different entrainers were used for the separation process--dimethyl sulfoxide (DMSO), ethylene glycol (EG) and benzene--to test their viability for the acetone-chloro- form system. Thermodynamic feasibility analysis was done through ternary map diagrams. Two different thermody- namic models, NRTL and UNIQUAC, were explored for the study of overall process. Key words: Extractive distillation, Maximum boiling azeotrope, Relative volatility, Residue curve map, Distillation boundary 1. Introduction that pressure swing distillation is unattractive for the separation of acetone-chloroform system since its composition is not sensitive to The distillation process is based on the difference in the volatil- pressure changes [6]. ity of the components to be separated. However, for mixtures where azeotropes are present, conventional distillation have shown to be 2. Problem design and specification unable to effect the separation desired [1]. -

Gas Compatibility | Specialty Gases | Air Liquide
Gas Compatibility The compatibility data shown on the following pages has been compiled to assist in evaluating the appropriate materials to use in handling various gases. Prepared for use with dry (anhydrous) gases at a normal operating temperature of 70°F (21°C), information may vary if different operating conditions exist. It is extremely important that all gas control equipment be compatible with the gas being passed through it. The use of a device that is not compatible with the service gas may damage the unit and cause a leak that could result in property damage or personal injury. To eliminate potentially dangerous situations, always check for compatibility of materials Gas Encyclopaedia before using any gases in your gas control equipment. Systems and equipment used in oxidizer gas service (i.e. oxygen or nitrous oxide) must be cleaned for oxidizer service. First published in 1976, this reference Since combinations of gases are unlimited, mixtures (except for ethylene oxide/ halocarbon book quickly became the must- have and ethylene oxide/CO 2 sterilizing gas mixtures) are not listed in this Compat ibility Chart. in the gas industry. Over 1,000 pages Before using a gas mixture or any gas not listed in the chart, please contact your Air Liquide includes information such as thermo - representative for more information . dy namics, safety, tables of physical and biological properties, and much more. Locate the gas you are using in the first column. Available now in print or online at Compare the materials of construction for the equipment Directions www.airliquide.com – contact your you intend to use with the materials of construction shown in Air Liquide representative for more the Compatibility Chart. -

Acetone Safety Data Sheet According to Federal Register / Vol
Acetone Safety Data Sheet according to Federal Register / Vol. 77, No. 58 / Monday, March 26, 2012 / Rules and Regulations Date of issue: 11/12/1998 Revision date: 04/24/2018 Supersedes: 04/24/2018 Version: 1.3 SECTION 1: Identification 1.1. Identification Product form : Substance Substance name : Acetone Chemical name : 2-Propanone CAS-No. : 67-64-1 Product code : LC10420, LC10425 Formula : C3H6O Synonyms : 2-propanone / beta-ketopropane / dimethyl formaldehyde / dimethyl ketone / dimethylketal / DMK (=dimethyl ketone) / keto propane / methyl ketone / pyroacetic acid / pyroacetic ether / pyroacetic spirit 1.2. Recommended use and restrictions on use Use of the substance/mixture : Solvent Cleaning product Chemical raw material Recommended use : Laboratory chemicals Restrictions on use : Not for food, drug or household use 1.3. Supplier LabChem, Inc. Jackson's Pointe Commerce Park Building 1000, 1010 Jackson's Pointe Court Zelienople, PA 16063 - USA T 412-826-5230 - F 724-473-0647 1.4. Emergency telephone number Emergency number : CHEMTREC: 1-800-424-9300 or +1-703-741-5970 SECTION 2: Hazard(s) identification 2.1. Classification of the substance or mixture GHS-US classification Flammable liquids H225 Highly flammable liquid and vapour Category 2 Serious eye damage/eye H319 Causes serious eye irritation irritation Category 2A Specific target organ H336 May cause drowsiness or dizziness toxicity (single exposure) Category 3 Full text of H statements : see section 16 2.2. GHS Label elements, including precautionary statements GHS US labeling Hazard pictograms (GHS US) : GHS02 GHS07 Signal word (GHS US) : Danger Hazard statements (GHS US) : H225 - Highly flammable liquid and vapour H319 - Causes serious eye irritation H336 - May cause drowsiness or dizziness Precautionary statements (GHS US) : P210 - Keep away from heat, hot surfaces, open flames, sparks. -

INCOMPATIBLE CHEMICALS Up-Dated October 2011
INCOMPATIBLE CHEMICALS Up-dated October 2011 Sources Accident Prevention Manual for Industrial Operations, 6th ed., National Safety Council, Fire Protection Guide on Hazardous Materials, 6th ed., National Fire Protection Association; 49CFR173; recent laboratory inspections. Incompatible materials should not be stored together where they can be inadvertently mixed or where a spill or leak can cause danger. General guidelines are: 1. Oxygen and fuels must not be stored together. 2. Water reactive materials are not to be stored with flammables (except where a flammable is used to blanket a material such as sodium and then at least practical quantity), or in an area where they could become wet (under a sink, sprinkler head, shower, etc.) 3. Strong acids and bases are not to be stored together. 4. Materials which can produce poisonous gases must not be stored with products which accelerate the release of the gas. (Examples: cyanogens are not to be stored with an acid, or cleaning products containing chlorine are not to be stored with ammonia.) 5. Explosives (picric acid, etc.) are not to be stored with fuels. 6. Incompatible acids must not be stored together. (Examples: perchloric acid is not to be stored with a reducing agent such as sulfuric acid, as upon mixing, this could produce a shock sensitive explosive; nitric acid and acetic acid, a potential explosive mixture, must not be stored together.) Specific examples of incompatible items likely to be found in laboratories are: Chemical Store Away From or Out of Contact With Acetic Acid Chromic acid, nitric acid, hydroxyl compounds, ethylene glycol, perchloric acid, peroxides and permanganates. -

Intermolecular Forces, Liquids, Solids IM Forces and Physical Properties
Intermolecular Forces, Liquids, Solids Interactions Between Molecules: What does it take to separate two (or more) molecules from one another? -or- What holds molecules close to one another? Structure/Property Relationships Name Butane Acetone Isopropyl Alcohol Molecular Formula C4H10 C3H6O C3H8O Molar Mass 58 g/mol 58 g/mol 60 g/mol H Structure H H H H H O H H O H HCCCCH HCCCH HCCCH H H H H H H H H H H Boiling Point -0.6 oC 56 oC 82 oC Why is there this discrepancy in boiling point? INTERMOLECULAR FORCES! 1 Isopropyl alcohol > acetone > butane IM Forces and Physical Properties Why this difference in bp? Let's take a closer look at these molecules: Butane Acetone Isopropyl Alcohol Boiling Point -0.6 oC 56 oC 82 oC O OH Structure Nonpolar Bonds Present Polar Bonds Present Molecular Polarity Apparently, nonpolar molecules have weaker intermolecular forces. Why? 2 1 Electrostatic interactions of molecules Interactions that result from charges! • Governed by Coulomb's Law: nene Force d2 • Types of electrostatic "species" possible: 1. Ions- 2. Dipoles- 3. Induced Dipoles- 3 Possible Types of IM Interactions aka “van de Waals forces” 1. Ion-Dipole Interactions – Remember back to discussion of water as a solvent: Water hydrates an ionic solute. - + - + - + 2. Dipole-Dipole Interactions Interactions of + end of one dipole with the - end of another. O - + C H3C CH3 4 2 Possible Types of IM Interactions 3. Dipole-Induced Dipole Interactions 4. Induced Dipole-Induced Dipole Interactions – “Dispersion Forces“ In one instant: Nanoseconds later: – Aka London Forces – Present in ALL molecules! + - + - - + + - - - - ---- -- -- - - -- - - -- -- -- - - -- - ---- - -- --- - - --- Attractive Repulsive 5 Hydrogen Bonding Specialized case of dipole-dipole interaction • Occurs b/w 1.