Comprehensive Identification of Nuclear and Cytoplasmic TNRC6A-Associating Proteins
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transcriptomic Profiling of Equine and Viral Genes in Peripheral Blood
pathogens Article Transcriptomic Profiling of Equine and Viral Genes in Peripheral Blood Mononuclear Cells in Horses during Equine Herpesvirus 1 Infection Lila M. Zarski 1, Patty Sue D. Weber 2, Yao Lee 1 and Gisela Soboll Hussey 1,* 1 Department of Pathobiology and Diagnostic Investigation, Michigan State University, East Lansing, MI 48824, USA; [email protected] (L.M.Z.); [email protected] (Y.L.) 2 Department of Large Animal Clinical Sciences, Michigan State University, East Lansing, MI 48824, USA; [email protected] * Correspondence: [email protected] Abstract: Equine herpesvirus 1 (EHV-1) affects horses worldwide and causes respiratory dis- ease, abortions, and equine herpesvirus myeloencephalopathy (EHM). Following infection, a cell- associated viremia is established in the peripheral blood mononuclear cells (PBMCs). This viremia is essential for transport of EHV-1 to secondary infection sites where subsequent immunopathol- ogy results in diseases such as abortion or EHM. Because of the central role of PBMCs in EHV-1 pathogenesis, our goal was to establish a gene expression analysis of host and equine herpesvirus genes during EHV-1 viremia using RNA sequencing. When comparing transcriptomes of PBMCs during peak viremia to those prior to EHV-1 infection, we found 51 differentially expressed equine genes (48 upregulated and 3 downregulated). After gene ontology analysis, processes such as the interferon defense response, response to chemokines, the complement protein activation cascade, cell adhesion, and coagulation were overrepresented during viremia. Additionally, transcripts for EHV-1, EHV-2, and EHV-5 were identified in pre- and post-EHV-1-infection samples. Looking at Citation: Zarski, L.M.; Weber, P.S.D.; micro RNAs (miRNAs), 278 known equine miRNAs and 855 potentially novel equine miRNAs were Lee, Y.; Soboll Hussey, G. -

Autism Multiplex Family with 16P11.2P12.2 Microduplication Syndrome in Monozygotic Twins and Distal 16P11.2 Deletion in Their Brother
European Journal of Human Genetics (2012) 20, 540–546 & 2012 Macmillan Publishers Limited All rights reserved 1018-4813/12 www.nature.com/ejhg ARTICLE Autism multiplex family with 16p11.2p12.2 microduplication syndrome in monozygotic twins and distal 16p11.2 deletion in their brother Anne-Claude Tabet1,2,3,4, Marion Pilorge2,3,4, Richard Delorme5,6,Fre´de´rique Amsellem5,6, Jean-Marc Pinard7, Marion Leboyer6,8,9, Alain Verloes10, Brigitte Benzacken1,11,12 and Catalina Betancur*,2,3,4 The pericentromeric region of chromosome 16p is rich in segmental duplications that predispose to rearrangements through non-allelic homologous recombination. Several recurrent copy number variations have been described recently in chromosome 16p. 16p11.2 rearrangements (29.5–30.1 Mb) are associated with autism, intellectual disability (ID) and other neurodevelopmental disorders. Another recognizable but less common microdeletion syndrome in 16p11.2p12.2 (21.4 to 28.5–30.1 Mb) has been described in six individuals with ID, whereas apparently reciprocal duplications, studied by standard cytogenetic and fluorescence in situ hybridization techniques, have been reported in three patients with autism spectrum disorders. Here, we report a multiplex family with three boys affected with autism, including two monozygotic twins carrying a de novo 16p11.2p12.2 duplication of 8.95 Mb (21.28–30.23 Mb) characterized by single-nucleotide polymorphism array, encompassing both the 16p11.2 and 16p11.2p12.2 regions. The twins exhibited autism, severe ID, and dysmorphic features, including a triangular face, deep-set eyes, large and prominent nasal bridge, and tall, slender build. The eldest brother presented with autism, mild ID, early-onset obesity and normal craniofacial features, and carried a smaller, overlapping 16p11.2 microdeletion of 847 kb (28.40–29.25 Mb), inherited from his apparently healthy father. -

Dissertation
Regulation of gene silencing: From microRNA biogenesis to post-translational modifications of TNRC6 complexes DISSERTATION zur Erlangung des DOKTORGRADES DER NATURWISSENSCHAFTEN (Dr. rer. nat.) der Fakultät Biologie und Vorklinische Medizin der Universität Regensburg vorgelegt von Johannes Danner aus Eggenfelden im Jahr 2017 Das Promotionsgesuch wurde eingereicht am: 12.09.2017 Die Arbeit wurde angeleitet von: Prof. Dr. Gunter Meister Johannes Danner Summary ‘From microRNA biogenesis to post-translational modifications of TNRC6 complexes’ summarizes the two main projects, beginning with the influence of specific RNA binding proteins on miRNA biogenesis processes. The fate of the mature miRNA is determined by the incorporation into Argonaute proteins followed by a complex formation with TNRC6 proteins as core molecules of gene silencing complexes. miRNAs are transcribed as stem-loop structured primary transcripts (pri-miRNA) by Pol II. The further nuclear processing is carried out by the microprocessor complex containing the RNase III enzyme Drosha, which cleaves the pri-miRNA to precursor-miRNA (pre-miRNA). After Exportin-5 mediated transport of the pre-miRNA to the cytoplasm, the RNase III enzyme Dicer cleaves off the terminal loop resulting in a 21-24 nt long double-stranded RNA. One of the strands is incorporated in the RNA-induced silencing complex (RISC), where it directly interacts with a member of the Argonaute protein family. The miRNA guides the mature RISC complex to partially complementary target sites on mRNAs leading to gene silencing. During this process TNRC6 proteins interact with Argonaute and recruit additional factors to mediate translational repression and target mRNA destabilization through deadenylation and decapping leading to mRNA decay. -
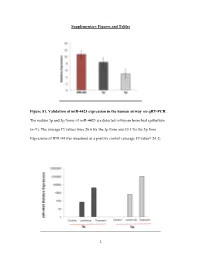
1 Supplementary Figures and Tables Figure S1. Validation
Supplementary Figures and Tables Figure S1. Validation of miR-4423 expression in the human airway via qRT-PCR. The mature 3p and 5p forms of miR-4423 are detected in human bronchial epithelium (n=9). The average Ct values were 26.6 for the 3p form and 30.1 for the 5p form. Expression of RNU44 was measured as a positive control (average Ct value= 24.3). 1 Figure S2. MiR-4423 overexpression. A) Stable and transient overexpression of a plasmid containing the miR-4423 precursor plus 200 bps of flanking region, results in the production of processed 3p and 5p forms. An empty vector was used as a control. Figure S3. Validation of miR-4423 biogenesis. A) Transfection with a siRNA targeting Dicer decreases Dicer mRNA levels in H1299 cells stably overexpressing miR-4423 (P=0.0001)(n=3). B) Levels of processed miR-4423-5p, miR-4423-3p, miR-10b, miR-21 and miR-26 are significantly decreased in Dicer knocked-down cells compared to control (P= 0.0014, P= 0.0013, P=0.04, P=0.01; P=0.019, respectively). Error bars indicate standard error and P values were determined using Student’s t test. 2 Figure S4. RNA-Seq profile of the miR-4423 locus using reads that associated with the AGO complex. Using a small RNA sequencing dataset from Hafner et al (1), one read that associated with TNRC6C and two reads that associated with AGO3 aligned uniquely to the 5p form while one read that associated with TNRC6A aligned uniquely to the 3p form. -

Circrna Circ 102049 Implicates in Pancreatic Ductal Adenocarcinoma Progression Through Activating CD80 by Targeting Mir-455-3P
Hindawi Mediators of Inflammation Volume 2021, Article ID 8819990, 30 pages https://doi.org/10.1155/2021/8819990 Research Article circRNA circ_102049 Implicates in Pancreatic Ductal Adenocarcinoma Progression through Activating CD80 by Targeting miR-455-3p Jie Zhu,1 Yong Zhou,1 Shanshan Zhu,1 Fei Li,1 Jiajia Xu,1 Liming Zhang,1 and Hairong Shu 2 1Medical Laboratory, Taizhou Central Hospital (Taizhou University Hospital), Taizhou, Zhejiang, China 2Department of Medical Service, Taizhou Central Hospital (Taizhou University Hospital), Taizhou, Zhejiang, China Correspondence should be addressed to Hairong Shu; [email protected] Received 30 September 2020; Revised 27 November 2020; Accepted 13 December 2020; Published 7 January 2021 Academic Editor: Xiaolu Jin Copyright © 2021 Jie Zhu et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Emerging evidence has shown that circular RNAs (circRNAs) and DNA methylation play important roles in the causation and progression of cancers. However, the roles of circRNAs and abnormal methylation genes in the tumorigenesis of pancreatic ductal adenocarcinoma (PDAC) are still largely unknown. Expression profiles of circRNA, gene methylation, and mRNA were downloaded from the GEO database, and differentially expressed genes were obtained via GEO2R, and a ceRNA network was constructed based on circRNA-miRNA pairs and miRNA-mRNA pairs. Inflammation-associated genes were collected from the GeneCards database. Then, functional enrichment analysis and protein-protein interaction (PPI) networks of inflammation- associated methylated expressed genes were investigated using Metascape and STRING databases, respectively, and visualized in Cytoscape. -

393LN V 393P 344SQ V 393P Probe Set Entrez Gene
393LN v 393P 344SQ v 393P Entrez fold fold probe set Gene Gene Symbol Gene cluster Gene Title p-value change p-value change chemokine (C-C motif) ligand 21b /// chemokine (C-C motif) ligand 21a /// chemokine (C-C motif) ligand 21c 1419426_s_at 18829 /// Ccl21b /// Ccl2 1 - up 393 LN only (leucine) 0.0047 9.199837 0.45212 6.847887 nuclear factor of activated T-cells, cytoplasmic, calcineurin- 1447085_s_at 18018 Nfatc1 1 - up 393 LN only dependent 1 0.009048 12.065 0.13718 4.81 RIKEN cDNA 1453647_at 78668 9530059J11Rik1 - up 393 LN only 9530059J11 gene 0.002208 5.482897 0.27642 3.45171 transient receptor potential cation channel, subfamily 1457164_at 277328 Trpa1 1 - up 393 LN only A, member 1 0.000111 9.180344 0.01771 3.048114 regulating synaptic membrane 1422809_at 116838 Rims2 1 - up 393 LN only exocytosis 2 0.001891 8.560424 0.13159 2.980501 glial cell line derived neurotrophic factor family receptor alpha 1433716_x_at 14586 Gfra2 1 - up 393 LN only 2 0.006868 30.88736 0.01066 2.811211 1446936_at --- --- 1 - up 393 LN only --- 0.007695 6.373955 0.11733 2.480287 zinc finger protein 1438742_at 320683 Zfp629 1 - up 393 LN only 629 0.002644 5.231855 0.38124 2.377016 phospholipase A2, 1426019_at 18786 Plaa 1 - up 393 LN only activating protein 0.008657 6.2364 0.12336 2.262117 1445314_at 14009 Etv1 1 - up 393 LN only ets variant gene 1 0.007224 3.643646 0.36434 2.01989 ciliary rootlet coiled- 1427338_at 230872 Crocc 1 - up 393 LN only coil, rootletin 0.002482 7.783242 0.49977 1.794171 expressed sequence 1436585_at 99463 BB182297 1 - up 393 -

Analyzing the Mirna-Gene Networks to Mine the Important Mirnas Under Skin of Human and Mouse
Hindawi Publishing Corporation BioMed Research International Volume 2016, Article ID 5469371, 9 pages http://dx.doi.org/10.1155/2016/5469371 Research Article Analyzing the miRNA-Gene Networks to Mine the Important miRNAs under Skin of Human and Mouse Jianghong Wu,1,2,3,4,5 Husile Gong,1,2 Yongsheng Bai,5,6 and Wenguang Zhang1 1 College of Animal Science, Inner Mongolia Agricultural University, Hohhot 010018, China 2Inner Mongolia Academy of Agricultural & Animal Husbandry Sciences, Hohhot 010031, China 3Inner Mongolia Prataculture Research Center, Chinese Academy of Science, Hohhot 010031, China 4State Key Laboratory of Genetic Resources and Evolution, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming 650223, China 5Department of Biology, Indiana State University, Terre Haute, IN 47809, USA 6The Center for Genomic Advocacy, Indiana State University, Terre Haute, IN 47809, USA Correspondence should be addressed to Yongsheng Bai; [email protected] and Wenguang Zhang; [email protected] Received 11 April 2016; Revised 15 July 2016; Accepted 27 July 2016 Academic Editor: Nicola Cirillo Copyright © 2016 Jianghong Wu et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Genetic networks provide new mechanistic insights into the diversity of species morphology. In this study, we have integrated the MGI, GEO, and miRNA database to analyze the genetic regulatory networks under morphology difference of integument of humans and mice. We found that the gene expression network in the skin is highly divergent between human and mouse. -

Identification of High-Confidence RNA Regulatory Elements By
Li et al. Genome Biology (2017) 18:169 DOI 10.1186/s13059-017-1298-8 METHOD Open Access Identification of high-confidence RNA regulatory elements by combinatorial classification of RNA–protein binding sites Yang Eric Li1†, Mu Xiao2†, Binbin Shi1†, Yu-Cheng T. Yang1, Dong Wang1, Fei Wang2, Marco Marcia3 and Zhi John Lu1* Abstract Crosslinking immunoprecipitation sequencing (CLIP-seq) technologies have enabled researchers to characterize transcriptome-wide binding sites of RNA-binding protein (RBP) with high resolution. We apply a soft-clustering method, RBPgroup, to various CLIP-seq datasets to group together RBPs that specifically bind the same RNA sites. Such combinatorial clustering of RBPs helps interpret CLIP-seq data and suggests functional RNA regulatory elements. Furthermore, we validate two RBP–RBP interactions in cell lines. Our approach links proteins and RNA motifs known to possess similar biochemical and cellular properties and can, when used in conjunction with additional experimental data, identify high-confidence RBP groups and their associated RNA regulatory elements. Keywords: RNA-binding protein, CLIP-seq, Non-negative matrix factorization, RBPgroup Background protein binding sites with high resolution in different RNA-binding proteins (RBPs) are essential to sustain mammalian cells [12–14]. The CLIP-seq data of mul- fundamental cellular functions, such as splicing, polya- tiple RNA binding proteins have been curated and denylation, transport, translation, and degradation of annotated in specific databases, such as CLIPdb, RNA transcripts [1, 2]. One study estimated that more POSTAR, and STARbase [15–17]. Several significant than 1500 different RBPs exist in human [3]. These studies improved the prediction of individual RBPs’ RBPs cooperate or compete with each other in binding binding sites by training on CLIP-seq and RNAcompete their RNA targets [4–6]. -
Control of the Localization and Function of a Mirna Silencing Component TNRC6A by Argonaute Protein
9856–9873 Nucleic Acids Research, 2015, Vol. 43, No. 20 Published online 7 October 2015 doi: 10.1093/nar/gkv1026 Control of the localization and function of a miRNA silencing component TNRC6A by Argonaute protein Kenji Nishi1, Tomoko Takahashi1, Masataka Suzawa1, Takuya Miyakawa2, Tatsuya Nagasawa1, Yvelt Ming3, Masaru Tanokura2 and Kumiko Ui-Tei1,3,* 1Department of Biological Sciences, Graduate School of Science, University of Tokyo, Tokyo 113-0033, Japan, 2Department of Applied Biological Chemistry, Graduate School of Agricultural and Life Sciences, University of Tokyo, Tokyo 113-8657, Japan and 3Department of Computational Biology and Medical Sciences, Graduate School of Frontier Sciences, University of Tokyo, Chiba-ken 277-8651, Japan Received April 14, 2015; Revised September 25, 2015; Accepted September 28, 2015 ABSTRACT complexes, leading to translational repression and mRNA degradation of miRNA-targeted mRNAs (4,11–17). GW182 family proteins play important roles in mi- In human HEp-2, HeLa, and many other cell lines, a croRNA (miRNA)-mediated RNA silencing. They di- human GW182 family protein, TNRC6A, is known to rectly interact with Argonaute (Ago) proteins in pro- be localized mainly in the processing (P) bodies (18–20), cessing bodies (P bodies), cytoplasmic foci involved which are cytoplasmic foci that contain proteins involved in in mRNA degradation and storage. Recently, we re- mRNA degradation, storage, and translational repression vealed that a human GW182 family protein, TNRC6A, (reviewed in 21). However, recently we found a nuclear lo- is a nuclear-cytoplasmic shuttling protein, and its calization signal (NLS) and a nuclear export signal (NES) subcellular localization is regulated by its own nu- in the central region of TNRC6A and showed that it is a clear localization signal and nuclear export sig- nuclear–cytoplasmic shuttling protein and its subcellular nal. -

Genome-Scale Rnai on Living-Cell Microarrays Identifies Novel Regulators of Drosophila Melanogaster TORC1–S6K Pathway Signaling
Genome-scale RNAi on living-cell microarrays identifies novel regulators of Drosophila melanogaster TORC1–S6K pathway signaling The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Lindquist, R. A. et al. “Genome-scale RNAi on Living-cell Microarrays Identifies Novel Regulators of Drosophila Melanogaster TORC1-S6K Pathway Signaling.” Genome Research 21.3 (2011): 433– 446. Web. As Published http://dx.doi.org/10.1101/gr.111492.110 Publisher Cold Spring Harbor Laboratory Press Version Final published version Citable link http://hdl.handle.net/1721.1/74542 Terms of Use Creative Commons Attribution Non-Commercial Detailed Terms http://creativecommons.org/licenses/by-nc/3.0/ Downloaded from genome.cshlp.org on November 1, 2012 - Published by Cold Spring Harbor Laboratory Press Genome-scale RNAi on living-cell microarrays identifies novel regulators of Drosophila melanogaster TORC1−S6K pathway signaling Robert A. Lindquist, Kathleen A. Ottina, Douglas B. Wheeler, et al. Genome Res. 2011 21: 433-446 originally published online January 14, 2011 Access the most recent version at doi:10.1101/gr.111492.110 Supplemental http://genome.cshlp.org/content/suppl/2010/12/03/gr.111492.110.DC1.html Material References This article cites 57 articles, 19 of which can be accessed free at: http://genome.cshlp.org/content/21/3/433.full.html#ref-list-1 Article cited in: http://genome.cshlp.org/content/21/3/433.full.html#related-urls Creative This article is distributed exclusively by Cold Spring Harbor Laboratory Press Commons for the first six months after the full-issue publication date (see License http://genome.cshlp.org/site/misc/terms.xhtml). -

The DEAD-Box RNA-Binding Protein DDX6 Regulates Parental RNA Decay for Cellular Reprogramming to Pluripotency
RESEARCH ARTICLE The DEAD-box RNA-binding protein DDX6 regulates parental RNA decay for cellular reprogramming to pluripotency Daisuke Kami1, Tomoya Kitani2, Akihiro Nakamura3, Naoki Wakui4, Rena Mizutani5, Masahito Ohue4, Fuyuki Kametani6, Nobuyoshi Akimitsu5, Satoshi Gojo1* 1 Department of Regenerative Medicine, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto, Japan, 2 Department of Cardiovascular Medicine, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto, Japan, 3 Department of Pediatric Cardiology and Nephrology, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto, Japan, a1111111111 4 Department of Computer Science, School of Computing, Tokyo Institute of Technology, Tokyo, Japan, a1111111111 5 Radioisotope Center, The University of Tokyo, Tokyo, Japan, 6 Department of Dementia and Higher Brain a1111111111 Function, Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan a1111111111 a1111111111 * [email protected] Abstract OPEN ACCESS Cellular transitions and differentiation processes require mRNAs supporting the new pheno- Citation: Kami D, Kitani T, Nakamura A, Wakui N, type but also the clearance of existing mRNAs for the parental phenotype. Cellular repro- Mizutani R, Ohue M, et al. (2018) The DEAD- gramming from fibroblasts to induced pluripotent stem cells (iPSCs) occurs at the early box RNA-binding protein DDX6 regulates parental stage of mesenchymal epithelial transition (MET) and involves drastic morphological RNA decay for cellular reprogramming to pluripotency. PLoS ONE 13(10): e0203708. https:// changes. We examined the molecular mechanism for MET, focusing on RNA metabolism. doi.org/10.1371/journal.pone.0203708 DDX6, an RNA helicase, was indispensable for iPSC formation, in addition to RO60 and Editor: Yoon Ki Kim, Korea University, REPUBLIC RNY1, a non-coding RNA, which form complexes involved in intracellular nucleotide sens- OF KOREA ing. -

A Network-Based Approach to Uncover Microrna-Mediated Disease Comorbidities and Potential Pathobiological Implications
www.nature.com/npjsba ARTICLE OPEN A network-based approach to uncover microRNA-mediated disease comorbidities and potential pathobiological implications Shuting Jin1,9, Xiangxiang Zeng2,9, Jiansong Fang3, Jiawei Lin1, Stephen Y. Chan4, Serpil C. Erzurum5,6 and Feixiong Cheng3,7,8* Disease–disease relationships (e.g., disease comorbidities) play crucial roles in pathobiological manifestations of diseases and personalized approaches to managing those conditions. In this study, we develop a network-based methodology, termed meta- path-based Disease Network (mpDisNet) capturing algorithm, to infer disease–disease relationships by assembling four biological networks: disease–miRNA, miRNA–gene, disease–gene, and the human protein–protein interactome. mpDisNet is a meta-path- based random walk to reconstruct the heterogeneous neighbors of a given node. mpDisNet uses a heterogeneous skip-gram model to solve the network representation of the nodes. We find that mpDisNet reveals high performance in inferring clinically reported disease–disease relationships, outperforming that of traditional gene/miRNA-overlap approaches. In addition, mpDisNet identifies network-based comorbidities for pulmonary diseases driven by underlying miRNA-mediated pathobiological pathways (i.e., hsa-let-7a- or hsa-let-7b-mediated airway epithelial apoptosis and pro-inflammatory cytokine pathways) as derived from the human interactome network analysis. The mpDisNet offers a powerful tool for network-based identification of disease–disease relationships with miRNA-mediated pathobiological pathways. 1234567890():,; npj Systems Biology and Applications (2019) ; https://doi.org/10.1038/s41540-019-0115-25:41 INTRODUCTION or completely pairing with their 3′ UTR region, thereby reducing The manifestation and clinical severity of human disease are the stability of the target miRNA or inhibiting translation to 9 affected by myriad factors, including genetic, epigenetic, lifestyle, downregulate the expression of genes of interest.