TNRC6 Proteins Modulate Hepatitis C Virus Replication by Spatially Regulating the Binding of Mir-122/Ago2 Complexes to Viral RNA You Li 1,2,*, Li Wang2,3,Efra´In E
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transcriptomic Profiling of Equine and Viral Genes in Peripheral Blood
pathogens Article Transcriptomic Profiling of Equine and Viral Genes in Peripheral Blood Mononuclear Cells in Horses during Equine Herpesvirus 1 Infection Lila M. Zarski 1, Patty Sue D. Weber 2, Yao Lee 1 and Gisela Soboll Hussey 1,* 1 Department of Pathobiology and Diagnostic Investigation, Michigan State University, East Lansing, MI 48824, USA; [email protected] (L.M.Z.); [email protected] (Y.L.) 2 Department of Large Animal Clinical Sciences, Michigan State University, East Lansing, MI 48824, USA; [email protected] * Correspondence: [email protected] Abstract: Equine herpesvirus 1 (EHV-1) affects horses worldwide and causes respiratory dis- ease, abortions, and equine herpesvirus myeloencephalopathy (EHM). Following infection, a cell- associated viremia is established in the peripheral blood mononuclear cells (PBMCs). This viremia is essential for transport of EHV-1 to secondary infection sites where subsequent immunopathol- ogy results in diseases such as abortion or EHM. Because of the central role of PBMCs in EHV-1 pathogenesis, our goal was to establish a gene expression analysis of host and equine herpesvirus genes during EHV-1 viremia using RNA sequencing. When comparing transcriptomes of PBMCs during peak viremia to those prior to EHV-1 infection, we found 51 differentially expressed equine genes (48 upregulated and 3 downregulated). After gene ontology analysis, processes such as the interferon defense response, response to chemokines, the complement protein activation cascade, cell adhesion, and coagulation were overrepresented during viremia. Additionally, transcripts for EHV-1, EHV-2, and EHV-5 were identified in pre- and post-EHV-1-infection samples. Looking at Citation: Zarski, L.M.; Weber, P.S.D.; micro RNAs (miRNAs), 278 known equine miRNAs and 855 potentially novel equine miRNAs were Lee, Y.; Soboll Hussey, G. -

Autism Multiplex Family with 16P11.2P12.2 Microduplication Syndrome in Monozygotic Twins and Distal 16P11.2 Deletion in Their Brother
European Journal of Human Genetics (2012) 20, 540–546 & 2012 Macmillan Publishers Limited All rights reserved 1018-4813/12 www.nature.com/ejhg ARTICLE Autism multiplex family with 16p11.2p12.2 microduplication syndrome in monozygotic twins and distal 16p11.2 deletion in their brother Anne-Claude Tabet1,2,3,4, Marion Pilorge2,3,4, Richard Delorme5,6,Fre´de´rique Amsellem5,6, Jean-Marc Pinard7, Marion Leboyer6,8,9, Alain Verloes10, Brigitte Benzacken1,11,12 and Catalina Betancur*,2,3,4 The pericentromeric region of chromosome 16p is rich in segmental duplications that predispose to rearrangements through non-allelic homologous recombination. Several recurrent copy number variations have been described recently in chromosome 16p. 16p11.2 rearrangements (29.5–30.1 Mb) are associated with autism, intellectual disability (ID) and other neurodevelopmental disorders. Another recognizable but less common microdeletion syndrome in 16p11.2p12.2 (21.4 to 28.5–30.1 Mb) has been described in six individuals with ID, whereas apparently reciprocal duplications, studied by standard cytogenetic and fluorescence in situ hybridization techniques, have been reported in three patients with autism spectrum disorders. Here, we report a multiplex family with three boys affected with autism, including two monozygotic twins carrying a de novo 16p11.2p12.2 duplication of 8.95 Mb (21.28–30.23 Mb) characterized by single-nucleotide polymorphism array, encompassing both the 16p11.2 and 16p11.2p12.2 regions. The twins exhibited autism, severe ID, and dysmorphic features, including a triangular face, deep-set eyes, large and prominent nasal bridge, and tall, slender build. The eldest brother presented with autism, mild ID, early-onset obesity and normal craniofacial features, and carried a smaller, overlapping 16p11.2 microdeletion of 847 kb (28.40–29.25 Mb), inherited from his apparently healthy father. -

Signature Redacted Signatureredacted
The biochemical basis for the cooperative action of microRNAs S ISTITUTE By By MAssACHUSEOFTECHNOLOGY > Daniel Briskin OCT 0 12019 B.S., Molecular Genetics (2012) LIBRARIES 0 Ohio State University SUBMITTED TO THE DEPARTMENT OF BIOLOGY IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY AT THE MASSACHUSETTS INSTITUTE OF TECHNOLOGY -SEPTEMBER-2019- -fve e 7-1012 © 2019 Massachusetts Institute of Technology All rights reserved Signature redacted Signature of Author: DanielBriskin Department of Biology 17 September 2019 Signature redacted Certified By:__ David P. Bartel Professor of Biology Thesis Supervisor Accepted By: Signatureredacted -V Stephen Bell Uncas and Helen Whitaker Professor of Biology Investigator, Howard Hughes Medical Institute Co-Director, Biology Graduate Committee 1 2 The biochemical basis for the cooperative action of microRNAs by Daniel Briskin Submitted to the Department of Biology on 17 September 2019 In partial fulfillment of the requirements for the degree of doctor of philosophy Abstract In metazoans, microRNAs (miRNAs) act to repress mRNAs through a combination of translational repression and target degradation. miRNAs predominantly pair within the 3' untranslated region (3' UTR) of the mRNA. In cells, closely spaced miRNA target sites within an mRNA can act cooperatively, leading to more repression of the target mRNA than expected by independent action at each site. This dissertation details the use of purified miRNA-AGO2 complexes, synthetic target RNAs, and a purified domain of TNRC6B that is able to simultaneously bind multiple AGO proteins. We examined the target site occupancy and affinities for miRNA-AGO2 binding in the absence and presence of TNRC6B, for target RNAs with a single miRNA site as well as multiple miRNA sites spaced at varying distances. -

Proquest Dissertations
INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand comer and continuing from left to right in equal sections with small overlaps. Each original is also photographed in one exposure and is included in reduced form at the back of the book. Photographs included in the original manuscript have been reproduced xerographically in this copy. Higher quality 6” x 9” black and white photographic prints are available for any photographs or illustrations appearing in this copy for an additional charge. Contact UMI directly to order. UMI Bell & Howell Information and Learning 300 North Zeeb Road, Ann Arbor, Ml 48106-1346 USA 800-521-0600 NOTE TO USERS Page(s) missing in number only; text follows. Microfilmed as received. 222-229 This reproduction is the best copy available. UMI MOLECULAR GENETIC AND BIOCHEMICAL STUDIES OF THE HUMAN AND MOUSE MHC COMPLEMENT GENE CLUSTERS DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Zhenyu Yang, M.S. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

Dissertation
Regulation of gene silencing: From microRNA biogenesis to post-translational modifications of TNRC6 complexes DISSERTATION zur Erlangung des DOKTORGRADES DER NATURWISSENSCHAFTEN (Dr. rer. nat.) der Fakultät Biologie und Vorklinische Medizin der Universität Regensburg vorgelegt von Johannes Danner aus Eggenfelden im Jahr 2017 Das Promotionsgesuch wurde eingereicht am: 12.09.2017 Die Arbeit wurde angeleitet von: Prof. Dr. Gunter Meister Johannes Danner Summary ‘From microRNA biogenesis to post-translational modifications of TNRC6 complexes’ summarizes the two main projects, beginning with the influence of specific RNA binding proteins on miRNA biogenesis processes. The fate of the mature miRNA is determined by the incorporation into Argonaute proteins followed by a complex formation with TNRC6 proteins as core molecules of gene silencing complexes. miRNAs are transcribed as stem-loop structured primary transcripts (pri-miRNA) by Pol II. The further nuclear processing is carried out by the microprocessor complex containing the RNase III enzyme Drosha, which cleaves the pri-miRNA to precursor-miRNA (pre-miRNA). After Exportin-5 mediated transport of the pre-miRNA to the cytoplasm, the RNase III enzyme Dicer cleaves off the terminal loop resulting in a 21-24 nt long double-stranded RNA. One of the strands is incorporated in the RNA-induced silencing complex (RISC), where it directly interacts with a member of the Argonaute protein family. The miRNA guides the mature RISC complex to partially complementary target sites on mRNAs leading to gene silencing. During this process TNRC6 proteins interact with Argonaute and recruit additional factors to mediate translational repression and target mRNA destabilization through deadenylation and decapping leading to mRNA decay. -

A Set of Regulatory Genes Co-Expressed in Embryonic Human Brain Is Implicated in Disrupted Speech Development
Molecular Psychiatry https://doi.org/10.1038/s41380-018-0020-x ARTICLE A set of regulatory genes co-expressed in embryonic human brain is implicated in disrupted speech development 1 1 1 2 3 Else Eising ● Amaia Carrion-Castillo ● Arianna Vino ● Edythe A. Strand ● Kathy J. Jakielski ● 4,5 6 7 8 9 Thomas S. Scerri ● Michael S. Hildebrand ● Richard Webster ● Alan Ma ● Bernard Mazoyer ● 1,10 4,5 6,11 6,12 13 Clyde Francks ● Melanie Bahlo ● Ingrid E. Scheffer ● Angela T. Morgan ● Lawrence D. Shriberg ● Simon E. Fisher 1,10 Received: 22 September 2017 / Revised: 3 December 2017 / Accepted: 2 January 2018 © The Author(s) 2018. This article is published with open access Abstract Genetic investigations of people with impaired development of spoken language provide windows into key aspects of human biology. Over 15 years after FOXP2 was identified, most speech and language impairments remain unexplained at the molecular level. We sequenced whole genomes of nineteen unrelated individuals diagnosed with childhood apraxia of speech, a rare disorder enriched for causative mutations of large effect. Where DNA was available from unaffected parents, CHD3 SETD1A WDR5 fi 1234567890();,: we discovered de novo mutations, implicating genes, including , and . In other probands, we identi ed novel loss-of-function variants affecting KAT6A, SETBP1, ZFHX4, TNRC6B and MKL2, regulatory genes with links to neurodevelopment. Several of the new candidates interact with each other or with known speech-related genes. Moreover, they show significant clustering within a single co-expression module of genes highly expressed during early human brain development. This study highlights gene regulatory pathways in the developing brain that may contribute to acquisition of proficient speech. -

Datasheet Blank Template
SAN TA C RUZ BI OTEC HNOL OG Y, INC . DOM3Z (B-12): sc-393141 BACKGROUND APPLICATIONS DOM3Z (dom-3 homolog Z), also known as NG6 or DOM3L, is a 396 amino DOM3Z (B-12) is recommended for detection of DOM3Z of mouse, rat and acid ubiquitously expressed protein belonging to the DOM3Z family. The gene human origin by Western Blotting (starting dilution 1:100, dilution range encoding DOM3Z maps to human chromosome 6 in the major histocompati - 1:100-1:1000), immunoprecipitation [1-2 µg per 100-500 µg of total protein bility complex (MHC) class III region. Chromosome 6 contains 170 million base (1 ml of cell lysate)], immunofluorescence (starting dilution 1:50, dilution pairs and comprises nearly 6% of the human genome. Deletion of a portion range 1:50-1:500), immunohistochemistry (including paraffin-embedded of the q arm of chromosome 6 is associated with early onset intestinal can - sections) (starting dilution 1:50, dilution range 1:50-1:500) and solid phase cer, suggesting the presence of a cancer susceptibility locus. Additionally, ELISA (starting dilution 1:30, dilution range 1:30-1:3000). Porphyria cutanea tarda, Parkinson’s disease and Stickler syndrome are all Suitable for use as control antibody for DOM3Z siRNA (h): sc-95321, DOM3Z associated with genes that map to chromosome 6. siRNA (m): sc-143143, DOM3Z shRNA Plasmid (h): sc-95321-SH, DOM3Z shRNA Plasmid (m): sc-143143-SH, DOM3Z shRNA (h) Lentiviral Particles: REFERENCES sc-95321-V and DOM3Z shRNA (m) Lentiviral Particles: sc-143143-V. 1. Brunner, H.G., et al . -

DNA Methylation Alterations in Blood Cells of Toddlers with Down Syndrome
G C A T T A C G G C A T genes Article DNA Methylation Alterations in Blood Cells of Toddlers with Down Syndrome Oxana Yu. Naumova 1,2,* , Rebecca Lipschutz 2, Sergey Yu. Rychkov 1, Olga V. Zhukova 1 and Elena L. Grigorenko 2,3,4,* 1 Vavilov Institute of General Genetics RAS, 119991 Moscow, Russia; [email protected] (S.Y.R.); [email protected] (O.V.Z.) 2 Department of Psychology, University of Houston, Houston, TX 77204, USA; [email protected] 3 Department of Psychology, Saint-Petersburg State University, 199034 Saint Petersburg, Russia 4 Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA * Correspondence: [email protected] or [email protected] (O.Y.N.); [email protected] (E.L.G.) Abstract: Recent research has provided evidence on genome-wide alterations in DNA methylation patterns due to trisomy 21, which have been detected in various tissues of individuals with Down syndrome (DS) across different developmental stages. Here, we report new data on the systematic genome-wide DNA methylation perturbations in blood cells of individuals with DS from a previously understudied age group—young children. We show that the study findings are highly consistent with those from the prior literature. In addition, utilizing relevant published data from two other developmental stages, neonatal and adult, we track a quasi-longitudinal trend in the DS-associated DNA methylation patterns as a systematic epigenomic destabilization with age. Citation: Naumova, O.Y.; Lipschutz, R.; Rychkov, S.Y.; Keywords: Down syndrome; infants and toddlers; trisomy 21; DNA methylation; Illumina 450K Zhukova, O.V.; Grigorenko, E.L. -
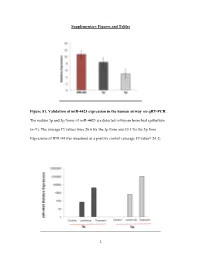
1 Supplementary Figures and Tables Figure S1. Validation
Supplementary Figures and Tables Figure S1. Validation of miR-4423 expression in the human airway via qRT-PCR. The mature 3p and 5p forms of miR-4423 are detected in human bronchial epithelium (n=9). The average Ct values were 26.6 for the 3p form and 30.1 for the 5p form. Expression of RNU44 was measured as a positive control (average Ct value= 24.3). 1 Figure S2. MiR-4423 overexpression. A) Stable and transient overexpression of a plasmid containing the miR-4423 precursor plus 200 bps of flanking region, results in the production of processed 3p and 5p forms. An empty vector was used as a control. Figure S3. Validation of miR-4423 biogenesis. A) Transfection with a siRNA targeting Dicer decreases Dicer mRNA levels in H1299 cells stably overexpressing miR-4423 (P=0.0001)(n=3). B) Levels of processed miR-4423-5p, miR-4423-3p, miR-10b, miR-21 and miR-26 are significantly decreased in Dicer knocked-down cells compared to control (P= 0.0014, P= 0.0013, P=0.04, P=0.01; P=0.019, respectively). Error bars indicate standard error and P values were determined using Student’s t test. 2 Figure S4. RNA-Seq profile of the miR-4423 locus using reads that associated with the AGO complex. Using a small RNA sequencing dataset from Hafner et al (1), one read that associated with TNRC6C and two reads that associated with AGO3 aligned uniquely to the 5p form while one read that associated with TNRC6A aligned uniquely to the 3p form. -

UNIVERSITY of CALIFORNIA Los Angeles the Subproteome Of
UNIVERSITY OF CALIFORNIA Los Angeles The subproteome of mitoBKCa channels from cardiomyocytes reveals novel insights into its mitochondrial import mechanism and function A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Molecular and Medical Pharmacology by Jin Zhang 2016 © Copyright by Jin Zhang 2016 ABSTRACT OF DISSERTATION The subproteome of mitoBKCa channels from cardiomyocytes reveals novel insights into its mitochondrial import mechanism and function by Jin Zhang Doctor of Philosophy in Molecular and Medical Pharmacology University of California, Los Angeles, 2016 Professor Ligia G Toro De Stefani, CO-Chair Professor Riccardo Olcese, CO-Chair BKCa channels are widely expressed ion channels and characterized by their large conductance to potassium and sensitivity to calcium and voltage. It is typically observed at the plasma membrane in the majority of cell types, with adult cardiomyocytes being an exception. Previously, both electrophysiological and immunological studies have detected BKCa channels in + cardiomyocytes mitoplasts, confirming the presence of this K channel (mitoBKCa); however, its physiological functions have not yet fully understood. This work is the first one to examine the interactome of mitoBKCa channels in adult heart (isolated cardiomyocytes and whole ventricle). We used a directed proteomic approach aided by co-immunoprecipitation with BKCa antibodies and pull-down with recombinant DEC sequences. ii As a result, we identified an extensive network of the mitoBKCa, channel and overall, a total of 1079 different proteins were identified as partners of the BKCa channel in cardiomyocytes and left ventricle, including 151 mitochondrial proteins. Two putative protein partners were selected to validate and further examine the associations with BKCa channels: i) the Tom22 from the mitochondrial import system, and ii) the adenine nucleotide translocator (ANT), which is linked to oxidative phosphorylation and the regulation of mPTP. -

Autism and Cancer Share Risk Genes, Pathways, and Drug Targets
TIGS 1255 No. of Pages 8 Forum Table 1 summarizes the characteristics of unclear whether this is related to its signal- Autism and Cancer risk genes for ASD that are also risk genes ing function or a consequence of a second for cancers, extending the original finding independent PTEN activity, but this dual Share Risk Genes, that the PI3K-Akt-mTOR signaling axis function may provide the rationale for the (involving PTEN, FMR1, NF1, TSC1, and dominant role of PTEN in cancer and Pathways, and Drug TSC2) was associated with inherited risk autism. Other genes encoding common Targets for both cancer and ASD [6–9]. Recent tumor signaling pathways include MET8[1_TD$IF],[2_TD$IF] genome-wide exome-sequencing studies PTK7, and HRAS, while p53, AKT, mTOR, Jacqueline N. Crawley,1,2,* of de novo variants in ASD and cancer WNT, NOTCH, and MAPK are compo- Wolf-Dietrich Heyer,3,4 and have begun to uncover considerable addi- nents of signaling pathways regulating Janine M. LaSalle1,4,5 tional overlap. What is surprising about the the nuclear factors described above. genes in Table 1 is not necessarily the Autism is a neurodevelopmental number of risk genes found in both autism Autism is comorbid with several mono- and cancer, but the shared functions of genic neurodevelopmental disorders, disorder, diagnosed behaviorally genes in chromatin remodeling and including Fragile X (FMR1), Rett syndrome by social and communication genome maintenance, transcription fac- (MECP2), Phelan-McDermid (SHANK3), fi de cits, repetitive behaviors, tors, and signal transduction pathways 15q duplication syndrome (UBE3A), and restricted interests. Recent leading to nuclear changes [7,8].