Rare Coding Variants and X-Linked Loci Associated with Age at Menarche
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transcriptomic Profiling of Equine and Viral Genes in Peripheral Blood
pathogens Article Transcriptomic Profiling of Equine and Viral Genes in Peripheral Blood Mononuclear Cells in Horses during Equine Herpesvirus 1 Infection Lila M. Zarski 1, Patty Sue D. Weber 2, Yao Lee 1 and Gisela Soboll Hussey 1,* 1 Department of Pathobiology and Diagnostic Investigation, Michigan State University, East Lansing, MI 48824, USA; [email protected] (L.M.Z.); [email protected] (Y.L.) 2 Department of Large Animal Clinical Sciences, Michigan State University, East Lansing, MI 48824, USA; [email protected] * Correspondence: [email protected] Abstract: Equine herpesvirus 1 (EHV-1) affects horses worldwide and causes respiratory dis- ease, abortions, and equine herpesvirus myeloencephalopathy (EHM). Following infection, a cell- associated viremia is established in the peripheral blood mononuclear cells (PBMCs). This viremia is essential for transport of EHV-1 to secondary infection sites where subsequent immunopathol- ogy results in diseases such as abortion or EHM. Because of the central role of PBMCs in EHV-1 pathogenesis, our goal was to establish a gene expression analysis of host and equine herpesvirus genes during EHV-1 viremia using RNA sequencing. When comparing transcriptomes of PBMCs during peak viremia to those prior to EHV-1 infection, we found 51 differentially expressed equine genes (48 upregulated and 3 downregulated). After gene ontology analysis, processes such as the interferon defense response, response to chemokines, the complement protein activation cascade, cell adhesion, and coagulation were overrepresented during viremia. Additionally, transcripts for EHV-1, EHV-2, and EHV-5 were identified in pre- and post-EHV-1-infection samples. Looking at Citation: Zarski, L.M.; Weber, P.S.D.; micro RNAs (miRNAs), 278 known equine miRNAs and 855 potentially novel equine miRNAs were Lee, Y.; Soboll Hussey, G. -

Autism Multiplex Family with 16P11.2P12.2 Microduplication Syndrome in Monozygotic Twins and Distal 16P11.2 Deletion in Their Brother
European Journal of Human Genetics (2012) 20, 540–546 & 2012 Macmillan Publishers Limited All rights reserved 1018-4813/12 www.nature.com/ejhg ARTICLE Autism multiplex family with 16p11.2p12.2 microduplication syndrome in monozygotic twins and distal 16p11.2 deletion in their brother Anne-Claude Tabet1,2,3,4, Marion Pilorge2,3,4, Richard Delorme5,6,Fre´de´rique Amsellem5,6, Jean-Marc Pinard7, Marion Leboyer6,8,9, Alain Verloes10, Brigitte Benzacken1,11,12 and Catalina Betancur*,2,3,4 The pericentromeric region of chromosome 16p is rich in segmental duplications that predispose to rearrangements through non-allelic homologous recombination. Several recurrent copy number variations have been described recently in chromosome 16p. 16p11.2 rearrangements (29.5–30.1 Mb) are associated with autism, intellectual disability (ID) and other neurodevelopmental disorders. Another recognizable but less common microdeletion syndrome in 16p11.2p12.2 (21.4 to 28.5–30.1 Mb) has been described in six individuals with ID, whereas apparently reciprocal duplications, studied by standard cytogenetic and fluorescence in situ hybridization techniques, have been reported in three patients with autism spectrum disorders. Here, we report a multiplex family with three boys affected with autism, including two monozygotic twins carrying a de novo 16p11.2p12.2 duplication of 8.95 Mb (21.28–30.23 Mb) characterized by single-nucleotide polymorphism array, encompassing both the 16p11.2 and 16p11.2p12.2 regions. The twins exhibited autism, severe ID, and dysmorphic features, including a triangular face, deep-set eyes, large and prominent nasal bridge, and tall, slender build. The eldest brother presented with autism, mild ID, early-onset obesity and normal craniofacial features, and carried a smaller, overlapping 16p11.2 microdeletion of 847 kb (28.40–29.25 Mb), inherited from his apparently healthy father. -

A Case of Congenital Central Hypothyroidism Caused by a Novel Variant (Gln1255ter) in IGSF1 Gene
Türkkahraman D et al. A Novel Variant in IGSF1 Gene CASE REPORT DO I: 10.4274/jcrpe.galenos.2020.2020.0149 J Clin Res Pediatr Endocrinol 2021;13(3):353-357 A Case of Congenital Central Hypothyroidism Caused by a Novel Variant (Gln1255Ter) in IGSF1 Gene Doğa Türkkahraman1, Nimet Karataş Torun2, Nadide Cemre Randa3 1University of Health Sciences Turkey, Antalya Training and Research Hospital, Clinic of Pediatric Endocrinology, Antalya, Turkey 2University of Healty Sciences Turkey, Antalya Training and Research Hospital, Clinic of Pediatrics, Antalya, Turkey 3University of Healty Sciences Turkey, Antalya Training and Research Hospital, Clinic of Medical Genetics, Antalya, Turkey What is already known on this topic? Mutations in the immunoglobulin superfamily, member 1 (IGSF1) gene that mainly regulates pituitary thyrotrope function lead to X-linked hypothyroidism characterized by congenital hypothyroidism of central origin and testicular enlargement. The clinical features associated with IGSF1 mutations are variable, but prolactin and/or growth hormone deficiency, and discordance between timing of testicular growth and rise of serum testosterone levels could be seen. What this study adds? Genetic analysis revealed a novel c.3763C>T variant in the IGSF1 gene. To our knowledge, this is the first reported case of IGSF1 deficiency from Turkey. Additionally, as in our case, early testicular enlargement but delayed testosterone rise should be evaluated in all boys with central hypothyroidism, as macro-orchidism is usually seen in adulthood. Abstract Loss-of-function mutations in the immunoglobulin superfamily, member 1 (IGSF1) gene cause X-linked central hypothyroidism, and therefore its mutation affects mainly males. Central hypothyroidism in males is the hallmark of the disorder, however some patients additionally present with hypoprolactinemia, transient and partial growth hormone deficiency, early/normal timing of testicular enlargement but delayed testosterone rise in puberty, and adult macro-orchidism. -

MECHANISMS in ENDOCRINOLOGY: Novel Genetic Causes of Short Stature
J M Wit and others Genetics of short stature 174:4 R145–R173 Review MECHANISMS IN ENDOCRINOLOGY Novel genetic causes of short stature 1 1 2 2 Jan M Wit , Wilma Oostdijk , Monique Losekoot , Hermine A van Duyvenvoorde , Correspondence Claudia A L Ruivenkamp2 and Sarina G Kant2 should be addressed to J M Wit Departments of 1Paediatrics and 2Clinical Genetics, Leiden University Medical Center, PO Box 9600, 2300 RC Leiden, Email The Netherlands [email protected] Abstract The fast technological development, particularly single nucleotide polymorphism array, array-comparative genomic hybridization, and whole exome sequencing, has led to the discovery of many novel genetic causes of growth failure. In this review we discuss a selection of these, according to a diagnostic classification centred on the epiphyseal growth plate. We successively discuss disorders in hormone signalling, paracrine factors, matrix molecules, intracellular pathways, and fundamental cellular processes, followed by chromosomal aberrations including copy number variants (CNVs) and imprinting disorders associated with short stature. Many novel causes of GH deficiency (GHD) as part of combined pituitary hormone deficiency have been uncovered. The most frequent genetic causes of isolated GHD are GH1 and GHRHR defects, but several novel causes have recently been found, such as GHSR, RNPC3, and IFT172 mutations. Besides well-defined causes of GH insensitivity (GHR, STAT5B, IGFALS, IGF1 defects), disorders of NFkB signalling, STAT3 and IGF2 have recently been discovered. Heterozygous IGF1R defects are a relatively frequent cause of prenatal and postnatal growth retardation. TRHA mutations cause a syndromic form of short stature with elevated T3/T4 ratio. Disorders of signalling of various paracrine factors (FGFs, BMPs, WNTs, PTHrP/IHH, and CNP/NPR2) or genetic defects affecting cartilage extracellular matrix usually cause disproportionate short stature. -

Dissertation
Regulation of gene silencing: From microRNA biogenesis to post-translational modifications of TNRC6 complexes DISSERTATION zur Erlangung des DOKTORGRADES DER NATURWISSENSCHAFTEN (Dr. rer. nat.) der Fakultät Biologie und Vorklinische Medizin der Universität Regensburg vorgelegt von Johannes Danner aus Eggenfelden im Jahr 2017 Das Promotionsgesuch wurde eingereicht am: 12.09.2017 Die Arbeit wurde angeleitet von: Prof. Dr. Gunter Meister Johannes Danner Summary ‘From microRNA biogenesis to post-translational modifications of TNRC6 complexes’ summarizes the two main projects, beginning with the influence of specific RNA binding proteins on miRNA biogenesis processes. The fate of the mature miRNA is determined by the incorporation into Argonaute proteins followed by a complex formation with TNRC6 proteins as core molecules of gene silencing complexes. miRNAs are transcribed as stem-loop structured primary transcripts (pri-miRNA) by Pol II. The further nuclear processing is carried out by the microprocessor complex containing the RNase III enzyme Drosha, which cleaves the pri-miRNA to precursor-miRNA (pre-miRNA). After Exportin-5 mediated transport of the pre-miRNA to the cytoplasm, the RNase III enzyme Dicer cleaves off the terminal loop resulting in a 21-24 nt long double-stranded RNA. One of the strands is incorporated in the RNA-induced silencing complex (RISC), where it directly interacts with a member of the Argonaute protein family. The miRNA guides the mature RISC complex to partially complementary target sites on mRNAs leading to gene silencing. During this process TNRC6 proteins interact with Argonaute and recruit additional factors to mediate translational repression and target mRNA destabilization through deadenylation and decapping leading to mRNA decay. -

Effects of Chronic Stress on Prefrontal Cortex Transcriptome in Mice Displaying Different Genetic Backgrounds
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Springer - Publisher Connector J Mol Neurosci (2013) 50:33–57 DOI 10.1007/s12031-012-9850-1 Effects of Chronic Stress on Prefrontal Cortex Transcriptome in Mice Displaying Different Genetic Backgrounds Pawel Lisowski & Marek Wieczorek & Joanna Goscik & Grzegorz R. Juszczak & Adrian M. Stankiewicz & Lech Zwierzchowski & Artur H. Swiergiel Received: 14 May 2012 /Accepted: 25 June 2012 /Published online: 27 July 2012 # The Author(s) 2012. This article is published with open access at Springerlink.com Abstract There is increasing evidence that depression signaling pathway (Clic6, Drd1a,andPpp1r1b). LA derives from the impact of environmental pressure on transcriptome affected by CMS was associated with genetically susceptible individuals. We analyzed the genes involved in behavioral response to stimulus effects of chronic mild stress (CMS) on prefrontal cor- (Fcer1g, Rasd2, S100a8, S100a9, Crhr1, Grm5,and tex transcriptome of two strains of mice bred for high Prkcc), immune effector processes (Fcer1g, Mpo,and (HA)and low (LA) swim stress-induced analgesia that Igh-VJ558), diacylglycerol binding (Rasgrp1, Dgke, differ in basal transcriptomic profiles and depression- Dgkg,andPrkcc), and long-term depression (Crhr1, like behaviors. We found that CMS affected 96 and 92 Grm5,andPrkcc) and/or coding elements of dendrites genes in HA and LA mice, respectively. Among genes (Crmp1, Cntnap4,andPrkcc) and myelin proteins with the same expression pattern in both strains after (Gpm6a, Mal,andMog). The results indicate significant CMS, we observed robust upregulation of Ttr gene contribution of genetic background to differences in coding transthyretin involved in amyloidosis, seizures, stress response gene expression in the mouse prefrontal stroke-like episodes, or dementia. -
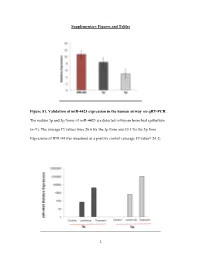
1 Supplementary Figures and Tables Figure S1. Validation
Supplementary Figures and Tables Figure S1. Validation of miR-4423 expression in the human airway via qRT-PCR. The mature 3p and 5p forms of miR-4423 are detected in human bronchial epithelium (n=9). The average Ct values were 26.6 for the 3p form and 30.1 for the 5p form. Expression of RNU44 was measured as a positive control (average Ct value= 24.3). 1 Figure S2. MiR-4423 overexpression. A) Stable and transient overexpression of a plasmid containing the miR-4423 precursor plus 200 bps of flanking region, results in the production of processed 3p and 5p forms. An empty vector was used as a control. Figure S3. Validation of miR-4423 biogenesis. A) Transfection with a siRNA targeting Dicer decreases Dicer mRNA levels in H1299 cells stably overexpressing miR-4423 (P=0.0001)(n=3). B) Levels of processed miR-4423-5p, miR-4423-3p, miR-10b, miR-21 and miR-26 are significantly decreased in Dicer knocked-down cells compared to control (P= 0.0014, P= 0.0013, P=0.04, P=0.01; P=0.019, respectively). Error bars indicate standard error and P values were determined using Student’s t test. 2 Figure S4. RNA-Seq profile of the miR-4423 locus using reads that associated with the AGO complex. Using a small RNA sequencing dataset from Hafner et al (1), one read that associated with TNRC6C and two reads that associated with AGO3 aligned uniquely to the 5p form while one read that associated with TNRC6A aligned uniquely to the 3p form. -

Circrna Circ 102049 Implicates in Pancreatic Ductal Adenocarcinoma Progression Through Activating CD80 by Targeting Mir-455-3P
Hindawi Mediators of Inflammation Volume 2021, Article ID 8819990, 30 pages https://doi.org/10.1155/2021/8819990 Research Article circRNA circ_102049 Implicates in Pancreatic Ductal Adenocarcinoma Progression through Activating CD80 by Targeting miR-455-3p Jie Zhu,1 Yong Zhou,1 Shanshan Zhu,1 Fei Li,1 Jiajia Xu,1 Liming Zhang,1 and Hairong Shu 2 1Medical Laboratory, Taizhou Central Hospital (Taizhou University Hospital), Taizhou, Zhejiang, China 2Department of Medical Service, Taizhou Central Hospital (Taizhou University Hospital), Taizhou, Zhejiang, China Correspondence should be addressed to Hairong Shu; [email protected] Received 30 September 2020; Revised 27 November 2020; Accepted 13 December 2020; Published 7 January 2021 Academic Editor: Xiaolu Jin Copyright © 2021 Jie Zhu et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Emerging evidence has shown that circular RNAs (circRNAs) and DNA methylation play important roles in the causation and progression of cancers. However, the roles of circRNAs and abnormal methylation genes in the tumorigenesis of pancreatic ductal adenocarcinoma (PDAC) are still largely unknown. Expression profiles of circRNA, gene methylation, and mRNA were downloaded from the GEO database, and differentially expressed genes were obtained via GEO2R, and a ceRNA network was constructed based on circRNA-miRNA pairs and miRNA-mRNA pairs. Inflammation-associated genes were collected from the GeneCards database. Then, functional enrichment analysis and protein-protein interaction (PPI) networks of inflammation- associated methylated expressed genes were investigated using Metascape and STRING databases, respectively, and visualized in Cytoscape. -

TAC3 Gene Products Regulate Brain and Digestive System Gene Expression in the Spotted Sea Bass (Lateolabrax Maculatus)
ORIGINAL RESEARCH published: 14 August 2019 doi: 10.3389/fendo.2019.00556 TAC3 Gene Products Regulate Brain and Digestive System Gene Expression in the Spotted Sea Bass (Lateolabrax maculatus) Zhanxiong Zhang, Haishen Wen, Yun Li, Qing Li, Wenjuan Li, Yangyang Zhou, Lingyu Wang, Yang Liu, Likang Lyu and Xin Qi* Key Laboratory of Mariculture, Ministry of Education, Ocean University of China, Qingdao, China Neurokinin B (NKB) is a member of the tachykinin (tac) family that plays important roles in mammalian growth by modulating prolactin (PRL) synthesis and secretion and causing contraction of the stomach and intestine. However, its potential role in regulating growth of teleosts is less clear. We aimed to explore the role that NKB plays in regulating fish growth using the spotted sea bass (Lateolabrax maculatus) as a model. In the present study, two tac3 and two tacr3 genes were identified in the spotted sea bass. Sequence analysis showed that two tac3 transcripts, tac3a and tac3b, encode four NKBs: NKBa-13, NKBa-10, NKBb-13, and NKBb-10. Expression analysis in different Edited by: tissues showed that both genes are highly expressed in the brain, stomach and intestine Vance L. Trudeau, University of Ottawa, Canada of the spotted sea bass. In situ hybridization indicated that the tac3a and tac3b mRNAs Reviewed by: are both localized in several brain regions, such as the telencephalon and hypothalamus, Berta Levavi-Sivan, and that tacr3a and tacr3b are localized in the intestinal villus and gastric gland. To Hebrew University of Jerusalem, Israel Satoshi Ogawa, investigate the potential role of NKBs in regulating growth, in vitro experiments were Monash University Malaysia, Malaysia performed to detect the effect of NKBs on growth-related gene expression in the brain *Correspondence: and brain-gut peptide (BGP)-related genes in the stomach and intestine. -

Potential Role in Controlling Fish Reproduction
Neurokinin Bs and neurokinin B receptors in zebrafish- potential role in controlling fish reproduction Jakob Birana, Ori Palevitcha, Shifra Ben-Dorb, and Berta Levavi-Sivana,1 aDepartment of Animal Sciences, The Robert H. Smith Faculty of Agriculture, Food, and Environment, Hebrew University of Jerusalem, Rehovot 76100, Israel; and bDepartment of Biological Services, Weizmann Institute of Science, Rehovot 76100, Israel Edited by John E. Halver, University of Washington, Seattle, WA, and approved April 27, 2012 (received for review December 2, 2011) The endocrine regulation of vertebrate reproduction is achieved by zebrafish (Danio rerio) as a model, was to examine the in- the coordinated actions of several peptide neurohormones, tachy- volvement of NKB in fish reproduction. kinin among them. To study the evolutionary conservation and NKB is a member of the tachykinin (TK) family of peptides. physiological functions of neurokinin B (NKB), we identified tachy- TKs are characterized by a common carboxyl-terminal amino acid kinin (tac) and tac receptor (NKBR) genes from many fish species, sequence of FXGLM-NH2 (where X is a hydrophobic residue), and cloned two cDNA forms from zebrafish. Phylogenetic analysis and include substance P, neurokinin A (NKA) and NKB, as well γ showed that piscine Tac3s and mammalian neurokinin genes arise as neuropeptide K, neuropeptide- , and hemokinin-1 (7). NKB is from one lineage. High identity was found among different fish the only TK synthesized from the preprotachykinin-B gene (8), species in the region encoding the NKB; all shared the common C- which is currently designated as TAC3 in mammals, except for terminal sequence. Although the piscine Tac3 gene encodes for two rodents, where it was named Tac2. -

Genetics of Combined Pituitary Hormone Deficiency: Roadmap Into the Genome Era
REVIEW Genetics of Combined Pituitary Hormone Deficiency: Roadmap into the Genome Era Qing Fang,* Akima S. George,* Michelle L. Brinkmeier, Amanda H. Mortensen, Downloaded from https://academic.oup.com/edrv/article-abstract/37/6/636/2691717 by AAG Fac de Agronomia user on 21 October 2019 Peter Gergics, Leonard Y. M. Cheung, Alexandre Z. Daly, Adnan Ajmal, María Ines Pérez Millán, A. Bilge Ozel, Jacob O. Kitzman, Ryan E. Mills, Jun Z. Li, and Sally A. Camper Department of Human Genetics (Q.F., A.S.G., M.L.B., A.H.M., P.G., L.Y.M.C., A.Z.D., M.I.P.M., A.B.O., J.O.K., R.E.M., J.Z.L., S.A.C.), Graduate Program in Bioinformatics (A.S.G.), Endocrine Division, Department of Internal Medicine (A.A.), and Department of Computational Medicine and Bioinformatics (J.O.K., R.E.M., J.Z.L.), University of Michigan, Ann Arbor, Michigan 48109 The genetic basis for combined pituitary hormone deficiency (CPHD) is complex, involving 30 genes in a variety of syndromic and nonsyndromic presentations. Molecular diagnosis of this disorder is valuable for predicting disease progression, avoiding unnecessary surgery, and family planning. We expect that the application of high throughput sequencing will uncover additional contributing genes and eventually become a valuable tool for molecular diag- nosis. For example, in the last 3 years, six new genes have been implicated in CPHD using whole-exome sequencing. In this review, we present a historical perspective on gene discovery for CPHD and predict approaches that may facilitate future gene identification projects conducted by clinicians and basic scientists. -

Molecular Characterization of Selectively Vulnerable Neurons in Alzheimer’S Disease
RESOURCE https://doi.org/10.1038/s41593-020-00764-7 Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease Kun Leng1,2,3,4,13, Emmy Li1,2,3,13, Rana Eser 5, Antonia Piergies5, Rene Sit2, Michelle Tan2, Norma Neff 2, Song Hua Li5, Roberta Diehl Rodriguez6, Claudia Kimie Suemoto7,8, Renata Elaine Paraizo Leite7, Alexander J. Ehrenberg 5, Carlos A. Pasqualucci7, William W. Seeley5,9, Salvatore Spina5, Helmut Heinsen7,10, Lea T. Grinberg 5,7,11 ✉ and Martin Kampmann 1,2,12 ✉ Alzheimer’s disease (AD) is characterized by the selective vulnerability of specific neuronal populations, the molecular sig- natures of which are largely unknown. To identify and characterize selectively vulnerable neuronal populations, we used single-nucleus RNA sequencing to profile the caudal entorhinal cortex and the superior frontal gyrus—brain regions where neurofibrillary inclusions and neuronal loss occur early and late in AD, respectively—from postmortem brains spanning the progression of AD-type tau neurofibrillary pathology. We identified RORB as a marker of selectively vulnerable excitatory neu- rons in the entorhinal cortex and subsequently validated their depletion and selective susceptibility to neurofibrillary inclusions during disease progression using quantitative neuropathological methods. We also discovered an astrocyte subpopulation, likely representing reactive astrocytes, characterized by decreased expression of genes involved in homeostatic functions. Our characterization of selectively vulnerable neurons in AD paves the way for future mechanistic studies of selective vulnerability and potential therapeutic strategies for enhancing neuronal resilience. elective vulnerability is a fundamental feature of neurodegen- Here, we performed snRNA-seq on postmortem brain tissue erative diseases, in which different neuronal populations show from a cohort of individuals spanning the progression of AD-type a gradient of susceptibility to degeneration.