A Theoretical Study of the Relationship Between the Electrophilicity Ω Index and Hammett Constant Σp in [3+2] Cycloaddition Reactions of Aryl Azide/Alkyne Derivatives
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Brief Guide to the Nomenclature of Organic Chemistry
1 Brief Guide to the Nomenclature of Table 1: Components of the substitutive name Organic Chemistry (4S,5E)-4,6-dichlorohept-5-en-2-one for K.-H. Hellwich (Germany), R. M. Hartshorn (New Zealand), CH3 Cl O A. Yerin (Russia), T. Damhus (Denmark), A. T. Hutton (South 4 2 Africa). E-mail: [email protected] Sponsoring body: Cl 6 CH 5 3 IUPAC Division of Chemical Nomenclature and Structure suffix for principal hept(a) parent (heptane) one Representation. characteristic group en(e) unsaturation ending chloro substituent prefix 1 INTRODUCTION di multiplicative prefix S E stereodescriptors CHEMISTRY The universal adoption of an agreed nomenclature is a key tool for 2 4 5 6 locants ( ) enclosing marks efficient communication in the chemical sciences, in industry and Multiplicative prefixes (Table 2) are used when more than one for regulations associated with import/export or health and safety. fragment of a particular kind is present in a structure. Which kind of REPRESENTATION The International Union of Pure and Applied Chemistry (IUPAC) multiplicative prefix is used depends on the complexity of the provides recommendations on many aspects of nomenclature.1 The APPLIED corresponding fragment – e.g. trichloro, but tris(chloromethyl). basics of organic nomenclature are summarized here, and there are companion documents on the nomenclature of inorganic2 and Table 2: Multiplicative prefixes for simple/complicated entities polymer3 chemistry, with hyperlinks to original documents. An No. Simple Complicated No. Simple Complicated AND overall -

Singlet/Triplet State Anti/Aromaticity of Cyclopentadienylcation: Sensitivity to Substituent Effect
Article Singlet/Triplet State Anti/Aromaticity of CyclopentadienylCation: Sensitivity to Substituent Effect Milovan Stojanovi´c 1, Jovana Aleksi´c 1 and Marija Baranac-Stojanovi´c 2,* 1 Institute of Chemistry, Technology and Metallurgy, Center for Chemistry, University of Belgrade, Njegoševa 12, P.O. Box 173, 11000 Belgrade, Serbia; [email protected] (M.S.); [email protected] (J.A.) 2 Faculty of Chemistry, University of Belgrade, Studentski trg 12-16, P.O. Box 158, 11000 Belgrade, Serbia * Correspondence: [email protected]; Tel.: +381-11-3336741 Abstract: It is well known that singlet state aromaticity is quite insensitive to substituent effects, in the case of monosubstitution. In this work, we use density functional theory (DFT) calculations to examine the sensitivity of triplet state aromaticity to substituent effects. For this purpose, we chose the singlet state antiaromatic cyclopentadienyl cation, antiaromaticity of which reverses to triplet state aromaticity, conforming to Baird’s rule. The extent of (anti)aromaticity was evaluated by using structural (HOMA), magnetic (NICS), energetic (ISE), and electronic (EDDBp) criteria. We find that the extent of triplet state aromaticity of monosubstituted cyclopentadienyl cations is weaker than the singlet state aromaticity of benzene and is, thus, slightly more sensitive to substituent effects. As an addition to the existing literature data, we also discuss substituent effects on singlet state antiaromaticity of cyclopentadienyl cation. Citation: Stojanovi´c,M.; Aleksi´c,J.; Baranac-Stojanovi´c,M. Keywords: antiaromaticity; aromaticity; singlet state; triplet state; cyclopentadienyl cation; substituent Singlet/Triplet State effect Anti/Aromaticity of CyclopentadienylCation: Sensitivity to Substituent Effect. -

1971 Schreck, Nonlinear Hammett Relationships.Pdf
James 0. Schreck University of Northern Colorado Nonlinear Hammett Relationships Greeley, Colorado 80631 The Haminett equation is a well-known empirical relationship for correlating structure and re- activity. Specifically, it relates structure to both equilibrium and reaction rate constants for reactions of met* and para-substituted benzene derivatives. When the Hammett equation is written in the form log (k/ko) = .P (1) k and ko are the rate (or equilibrium) constants for the " reaction of the substituted and unsubstituted com- S!h$tttuent Cmrtant. 0 pounds, respectively; u is the substituent constant, and Figure 1. Types of lineor ond nonlinear Hammoll curves. p is the reaction constant. The substituent constant is a measure of the ability of the substitueut to change reaction such as the electron density at the reaction site and is indepen- dent of the reaction involved; whereas, the reaction con- stant is a measure of the sensitivity of the reaction series to changes in electron density at the reaction site. In shows how changes in structure which affect principally most cases the equation is valid only for substituents the second step can result in a nonlinear structure-re- in the meta- or para-positions of the benzene ring. (In activity correlation (6). In this case the rate control- a recent article, however, the relative chemical shifts ling step of the reaction changes from the first step to of OH in ortho-substituted phenols in DMSO are the second step due to the greater effect of changes in correlated excellently with ortho-substituents (I).) structure on the second step of the reaction. -

Nomenclature of Alkanes
Nomenclature of alkanes methane CH4 ethane CH3CH3 propane CH3CH2CH3 butane CH3CH2CH2CH3 pentane CH3CH2CH2CH2CH3 hexane CH3CH2CH2CH2CH2CH3 heptane CH3CH2CH2CH2CH2CH2CH3 octane CH3CH2CH2CH2CH2CH2CH2CH3 nonane CH3CH2CH2CH2CH2CH2CH2CH2CH3 decane CH3CH2CH2CH2CH2CH2CH2CH2CH2CH3 undecane CH3CH2CH2CH2CH2CH2CH2CH2CH2CH2CH3 dodecane CH3CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH3 Funky groups/alkanes iso- CH3 R isopropyl HC R CH3 isobutane/isobutyl R = CH3/CH2R (4 C’s) R isopentane/isopentyl R = CH2CH3/CH2CH2R (5 C’s) R isohexane/isohexyl R = CH2CH2CH3/CH2CH2R (6 C’s) R neo- CH3 neopentyl H3C C CH2 R R CH3 neopentane R = H (5 C’s) neohaxane/neohexyl R = CH3/CH2R (6 C’s) R 1° primary (n-) 2° secondary (sec-, s-) 3° tertiary (tert-, t-) H C H C H3C 3 3 CH CH2 CH CH2 CH CH2 H C CH H C CH H3C CH3 3 3 3 3 n- often used with strait chain compounds though it is not actually necessary. sec- sec-butyl H the substituent is attached s-butyl to a 2° C of butane (4 C’s) H3C CH2 C R CH3 no other "sec-" group tert- tert-butyl CH3 a substituent is attached to t-butyl the 3° C of a 4 C H3C C R molecule/unit CH3 tert-pentyl CH3 a substituent is attached to t-pentyl the 3° of a 5 C molecule/unit H3C CH2 C R CH3 no other "tert-" group Form of name #-followed by substituent name followed by parent hydrocarbon name • Determine longest continuous chain. o This is the parent hydrocarbon o If compound has two or more chains of the same length, parent hydrocarbon is chain with greatest number of substituents • Cite the name of substituent before the name of the parent hydrocarbon along with the number of the carbon to which it is attached--Substituents are listed in alphabetical order – neglecting prefixes such as di- tri- tert- etc. -

Marvin Charton - Correlation Analyst Par Excellence
Int. J. Mol. Sci. 2005, 6, 3-10 International Journal of Molecular Sciences ISSN 1422-0067 © 2005 by MDPI www.mdpi.org/ijms/ Marvin Charton - Correlation Analyst Par Excellence John Shorter 29A, Meadowfields, Whitby, North Yorkshire, YO21 1QF, UK. (Emeritus Reader in Chemistry, University of Hull), email: [email protected] Received: 10 September 2004 / Accepted: 19 October 2004 / Published: 31 January 2005 Abstract: Since the late 1950s Marvin Charton, physical organic chemist, of Pratt Institute, Brooklyn, New York has studied structure-property relationships through correlation analysis, and has become one of the leading authorities in this field. Keywords: Hammett equation, correlation analysis, linear free energy relationships, substituent effects and constants, biological activity. Introduction Marvin Charton, of Pratt Institute, Brooklyn, New York, describes his research interests in his CV as follows: "The study of structure-property quantitative relationships in chemistry and biology by means of correlation analysis. Applications of this work include chemical reaction mechanisms, the estimation of chemical and physical properties and chemical reactivities of chemical compounds, the design of bioactive molecules including medicinal drugs and pesticides, and the prediction of environmental toxicities and other properties." His first paper in this field appeared in 1958 and he has now published over 150 research papers and review articles. Marvin's forebears came to the USA from eastern Europe in the late 19th century. His surname may sound French, but his ancestry was Polish, and the relatives who came from Poland to America had the name of Charton. He sometimes suggests, half in fun, that one of his ancestors may have accompanied Napoleon from France to Moscow in 1812, but during the great retreat, by the time they reached Poland he was a little tired of marching, so he absented himself, and found a good place to live and a nice Polish girl to marry... -

And Nitro- Oxy-Polycyclic Aromatic Hydrocarbons
This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organization or the World Health Organization. Environmental Health Criteria 229 SELECTED NITRO- AND NITRO- OXY-POLYCYCLIC AROMATIC HYDROCARBONS First draft prepared by Drs J. Kielhorn, U. Wahnschaffe and I. Mangelsdorf, Fraunhofer Institute of Toxicology and Aerosol Research, Hanover, Germany Please note that the pagination and layout of this web version are not identical to the printed EHC Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organization and the World Health Organization, and produced within the framework of the Inter-Organization Programme for the Sound Management of Chemicals. The International Programme on Chemical Safety (IPCS), established in 1980, is a joint venture of the United Nations Environment Programme (UNEP), the International Labour Organization (ILO) and the World Health Organization (WHO). The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals. The Inter-Organization Programme for the Sound Management of Chemicals (IOMC) was established in 1995 by UNEP, ILO, the Food and Agriculture Organization of the United Nations, WHO, the United Nations Industrial Development Organization, the United Nations Institute for Training and Research and the Organisation for Economic Co- operation and Development (Participating Organizations), following recommendations made by the 1992 UN Conference on Environment and Development to strengthen cooperation and increase coordination in the field of chemical safety. -

II. Nomenclature Rules for Alkenes 1. the Parent Name Will Be the Longest
1 Lecture 9 II. Nomenclature Rules For Alkenes 1. The parent name will be the longest carbon chain that contains both carbons of the double bond. Drop the -ane suffix of the alkane name and add the –ene suffix. Never name the double bond as a prefix. If a double bond is present, you have an alkene, not an alkane. alkane + -ene = alkene 2. Begin numbering the chain at the end nearest the double bond. Always number through the double bond and identify its position in the longest chain with the lower number. In the older IUPAC rules the number for the double bond was placed in front of the stem name with a hyphen. Under the newer rules, the number for the double bond is placed right in front of “ene”, with hyphens. We will use the newer rules for specifying the location of pi bonds. 1 2 3456 H3CCHCH CH 2 CH2 CH3 hex-2-ene (newer rules) 2-hexene (older rules) 3. Indicate the position of any substituent group by the number of the carbon atom in the parent (longest) chain to which it is attached. CH 1 2 345 3 H3CCHCHCHCH2 CH CH3 6 7 CH3 Numbering is determined by the double bond, not the branches, because the double bond has 5,6-dimethylhept-3-ene (newer rules) higher priority than any alkyl branch. 5,6-dimethyl-3-heptene (older rules) 4. Number cycloalkenes so that the double bond is 1,2 (number through the double bond). Number in the direction about the ring so that the lowest number is used at the first point of difference. -
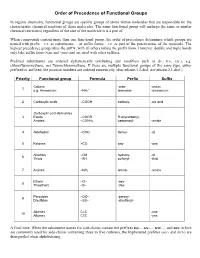
Priority of Functional Groups
Order of Precedence of Functional Groups In organic chemistry, functional groups are specific groups of atoms within molecules that are responsible for the characteristic chemical reactions of those molecules. The same functional group will undergo the same or similar chemical reaction(s) regardless of the size of the molecule it is a part of. When compounds contain more than one functional group, the order of precedence determines which groups are named with prefix – i.e. as substituents –, or suffix forms – i.e. as part of the parent name of the molecule. The highest precedence group takes the suffix, with all others taking the prefix form. However, double and triple bonds only take suffix form (-ene and -yne) and are used with other suffixes. Prefixed substituents are ordered alphabetically (excluding any modifiers such as di-, tri-, etc.), e.g. chlorofluoromethane, not fluorochloromethane. If there are multiple functional groups of the same type, either prefixed or suffixed, the position numbers are ordered numerically (thus ethane-1,2-diol, not ethane-2,1-diol.) Priority Functional group Formula Prefix Suffix Cations -onio- -onium 1 + e.g. Ammonium –NH4 ammonio- -ammonium 2 Carboxylic acids –COOH carboxy- -oic acid Carboxylic acid derivatives 3 Esters –COOR R-oxycarbonyl- Amides –CONH2 carbamoyl- -amide 4 Aldehydes –CHO formyl- -al 5 Ketones >CO oxo- -one Alcohols –OH hydroxy- -ol 6 Thiols –SH sulfanyl- -thiol 7 Amines –NH2 amino- -amine Ethers –O– -oxy- 8 Thioethers –S– -thio- Peroxides –OO– -peroxy- 9 Disulfides –SS– -disulfanyl- Alkenes C=C -ene 10 Alkynes C≡C -yne A final note: When the substituent names for side-chains contain the prefixes iso..., sec-..., tert-..., and neo (which are commonly used for side-chains containing three to five carbons), the hyphenated prefixes (sec- and tert-) are disregarded when alphabetizing. -

PHYSICAL ORGANIC Chemistryl
PHYSICAL ORGANIC CHEMISTRYl By EDWARD R. THORNTON Department of Chemistry, University of Pennsylvania, Philadelphia, Pennsylvania The author, who considers himself to have interests typical of a physical organic chemist, found about 2300 papers published during the last year that were of special interest to him. He confesses that he could not possibly read them all and hopes that those whose work may have been left out by neces sity or oversight will understand the problems involved. Most of this review discusses secondary and solvent isotope effects. Mention is also made of recent developments in quantum chemistry, es pecially in the qualitative discussion of orbital symmetry as a determining factor for certain reaction pathways. It cannot be claimed that enough space was available for comprehensive coverage of even these restricted topics. SECONDARY DEUTERIUM ISOTOPE EFFECTS A great deal of interesting information on secondary isotope effects has become available since the comprehensive review by Halevi (1) in 1963. Advances have been made in both experimental studies and theoretical un derstanding. Some of this new work, along with the present author's inter pretation of secondary isotope effects, will be discussed. Origin of secondary isotope effects.-A great deal of difficulty occurs in attempting to describe the causes of secondary isotope effects. Nevertheless, as far as the author knows, there is no case where an appropriate statistical treatment of isotope effects [Bigeleisen & Wolfsberg (2), Melander (3), Thornton (4)] does not adequately describe experiment. Even in terms of isotopic effects of the order of a few per cent, this theory and the approxima tions it uses are very precise. -

1 Chapter 3: Organic Compounds: Alkanes and Cycloalkanes
Chapter 3: Organic Compounds: Alkanes and Cycloalkanes >11 million organic compounds which are classified into families according to structure and reactivity Functional Group (FG): group of atoms which are part of a large molecule that have characteristic chemical behavior. FG’s behave similarly in every molecule they are part of. The chemistry of the organic molecule is defined by the function groups it contains 1 C C Alkanes Carbon - Carbon Multiple Bonds Carbon-heteroatom single bonds basic C N C C C X X= F, Cl, Br, I amines Alkenes Alkyl Halide H C C C O C C O Alkynes alcohols ethers acidic H H H C S C C C C S C C H sulfides C C thiols (disulfides) H H Arenes Carbonyl-oxygen double bonds (carbonyls) Carbon-nitrogen multiple bonds acidic basic O O O N H C H C O C Cl imine (Schiff base) aldehyde carboxylic acid acid chloride O O O O C C N C C C C O O C C nitrile (cyano group) ketones ester anhydrides O C N amide opsin Lys-NH2 + Lys- opsin H O H N rhodopsin H 2 Alkanes and Alkane Isomers Alkanes: organic compounds with only C-C and C-H single (s) bonds. general formula for alkanes: CnH(2n+2) Saturated hydrocarbons Hydrocarbons: contains only carbon and hydrogen Saturated" contains only single bonds Isomers: compounds with the same chemical formula, but different arrangement of atoms Constitutional isomer: have different connectivities (not limited to alkanes) C H O C4H10 C5H12 2 6 O OH butanol diethyl ether straight-chain or normal hydrocarbons branched hydrocarbons n-butane n-pentane Systematic Nomenclature (IUPAC System) Prefix-Parent-Suffix -

Physical Organic Chemistry
CHM 8304 Physical Organic Chemistry Thermodynamics and kinetics Outline: Isotope effects • see section 8.1 of A&D – experimental approach – primary isotope effect – secondary isotope effect – equilibrium isotope effect – solvent isotope effect – heavy atom isotope effects 2 Thermodynamics and kinetics 1 CHM 8304 Measurement of an isotope effect • performed to determine if a bond changes in a certain way during the rate-limiting step • expressed as a ratio whose numerator is the rate constant measured for the naturally abundant isotope and the denominator is the rate constant measured for the varied isotope – e.g. kH/kD 3 Types of isotope effects • kinetic isotope effects (kie): result from a change in the rate constant of a reaction : – normal effect: ratio > 1 – inverse effect: ratio < 1 – primary isotope effect: when the isotopically substituted bond is cleaved during the rate-limiting step – secondary isotope effect : attributable to a change of hybridation state, not cleavage of bonds • equilibrium isotope effects: result from displacement of an equilibrium 4 Thermodynamics and kinetics 2 CHM 8304 Origin of isotope effects • the origin of all isotope effects is a difference in the frequency of vibrational modes of a substituted molecule with respect to an unsubstituted molecule • it is these vibrational modes that principally affect the shape of the potential energy well on an energy surface 5 Zero point energy • zero point energy (ZPE) is the energy level of the vibrational ground state for most molecules at ambient temperature • -

Organic Chemistry Lecture Outline Chapter 16: Chemistry of Benzene: Electrophilic Aromatic Substitution
Organic Chemistry Lecture Outline Chapter 16: Chemistry of Benzene: Electrophilic Aromatic Substitution I. ELECTROPHILIC AROMATIC SUBSTITUTION (EAS) A. There are five general types of electrophilic aromatic substitution reactions. 1. Halogenation of benzene with Br2, Cl2 or I2 occurs through the same mechanism. a. Bromination of benzene occurs upon treatment with Br2 and FeBr3. b. Chlorination of benzene occurs upon treatment with Cl2 and FeCl3. c. Iodination of benzene occurs upon treatment with I2 and CuCl2. + 2. Nitration of benzene occurs through treatment with a nitronium ion, NO2. a. The nitronium ion can be generated from nitric acid and sulfuric acid. b. The nitronium ion can be generated from nitronium tetrafluoroborate. c. Nitration of benzene occurs through the same mechanism as halogenation. 3. Sulfonation of benzene occurs through treatment with fuming sulfuric acid. The reaction is reversible and products depend on the reaction conditions. a. In strong acid, sulfonation is favored. b. In hot, dilute acid, desulfonation is favored. c. Sulfonation of benzene occurs through the same mechanism as halogenation & nitration. 4. Alkylation (Friedel-Crafts Alkylation) of benzene involves substituting a hydrogen atom on a benzene ring with an alkyl group. This reaction occurs by treatment of benzene with a stable carbocation and aluminum trichloride. There are a number of problems with alkylation of an aromatic ring using the Friedel-Crafts reaction. a. Only alkyl halides can be used to generate the carbocation. b. Benzene rings substituted with electron-withdrawing groups are not reactive. c. The monosubstituted product initially formed is more reactive than the starting material d. Rearrangements can occur with unstable carbocations.