Singlet/Triplet State Anti/Aromaticity of Cyclopentadienylcation: Sensitivity to Substituent Effect
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Brief Guide to the Nomenclature of Organic Chemistry
1 Brief Guide to the Nomenclature of Table 1: Components of the substitutive name Organic Chemistry (4S,5E)-4,6-dichlorohept-5-en-2-one for K.-H. Hellwich (Germany), R. M. Hartshorn (New Zealand), CH3 Cl O A. Yerin (Russia), T. Damhus (Denmark), A. T. Hutton (South 4 2 Africa). E-mail: [email protected] Sponsoring body: Cl 6 CH 5 3 IUPAC Division of Chemical Nomenclature and Structure suffix for principal hept(a) parent (heptane) one Representation. characteristic group en(e) unsaturation ending chloro substituent prefix 1 INTRODUCTION di multiplicative prefix S E stereodescriptors CHEMISTRY The universal adoption of an agreed nomenclature is a key tool for 2 4 5 6 locants ( ) enclosing marks efficient communication in the chemical sciences, in industry and Multiplicative prefixes (Table 2) are used when more than one for regulations associated with import/export or health and safety. fragment of a particular kind is present in a structure. Which kind of REPRESENTATION The International Union of Pure and Applied Chemistry (IUPAC) multiplicative prefix is used depends on the complexity of the provides recommendations on many aspects of nomenclature.1 The APPLIED corresponding fragment – e.g. trichloro, but tris(chloromethyl). basics of organic nomenclature are summarized here, and there are companion documents on the nomenclature of inorganic2 and Table 2: Multiplicative prefixes for simple/complicated entities polymer3 chemistry, with hyperlinks to original documents. An No. Simple Complicated No. Simple Complicated AND overall -

Colour Structures in Supersymmetric QCD
Colour structures in Supersymmetric QCD Jorrit van den Boogaard August 23, 2010 In this thesis we will explain a novel method for calculating the colour struc- tures of Quantum ChromoDynamical processes, or QCD prosesses for short, with the eventual goal to apply such knowledge to Supersymmetric theory. QCD is the theory of the strong interactions, among quarks and gluons. From a Supersymmetric viewpoint, something we will explain in the first chapter, it is interesting to know as much as possible about QCD theory, for it will also tell us something about this Supersymmetric theory. Colour structures in particular, which give the strength of the strong force, can tell us something about the interactions with the Supersymmetric sector. There are a multitude of different ways to calculate these colour structures. The one described here will make use of diagrammatic methods and is therefore somewhat more user friendly. We therefore hope it will see more use in future calculations regarding these processes. We do warn the reader for the level of knowledge required to fully grasp the subject. Although this thesis is written with students with a bachelor degree in mind, one cannot escape touching somewhat dificult matterial. We have tried to explain things as much as possible in a way which should be clear to the reader. Sometimes however we saw it neccesary to skip some of the finer details of the subject. If one wants to learn more about these hiatuses we suggest looking at the material in the bibliography. For convenience we will use some common conventions in this thesis. -

Nomenclature of Alkanes
Nomenclature of alkanes methane CH4 ethane CH3CH3 propane CH3CH2CH3 butane CH3CH2CH2CH3 pentane CH3CH2CH2CH2CH3 hexane CH3CH2CH2CH2CH2CH3 heptane CH3CH2CH2CH2CH2CH2CH3 octane CH3CH2CH2CH2CH2CH2CH2CH3 nonane CH3CH2CH2CH2CH2CH2CH2CH2CH3 decane CH3CH2CH2CH2CH2CH2CH2CH2CH2CH3 undecane CH3CH2CH2CH2CH2CH2CH2CH2CH2CH2CH3 dodecane CH3CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH3 Funky groups/alkanes iso- CH3 R isopropyl HC R CH3 isobutane/isobutyl R = CH3/CH2R (4 C’s) R isopentane/isopentyl R = CH2CH3/CH2CH2R (5 C’s) R isohexane/isohexyl R = CH2CH2CH3/CH2CH2R (6 C’s) R neo- CH3 neopentyl H3C C CH2 R R CH3 neopentane R = H (5 C’s) neohaxane/neohexyl R = CH3/CH2R (6 C’s) R 1° primary (n-) 2° secondary (sec-, s-) 3° tertiary (tert-, t-) H C H C H3C 3 3 CH CH2 CH CH2 CH CH2 H C CH H C CH H3C CH3 3 3 3 3 n- often used with strait chain compounds though it is not actually necessary. sec- sec-butyl H the substituent is attached s-butyl to a 2° C of butane (4 C’s) H3C CH2 C R CH3 no other "sec-" group tert- tert-butyl CH3 a substituent is attached to t-butyl the 3° C of a 4 C H3C C R molecule/unit CH3 tert-pentyl CH3 a substituent is attached to t-pentyl the 3° of a 5 C molecule/unit H3C CH2 C R CH3 no other "tert-" group Form of name #-followed by substituent name followed by parent hydrocarbon name • Determine longest continuous chain. o This is the parent hydrocarbon o If compound has two or more chains of the same length, parent hydrocarbon is chain with greatest number of substituents • Cite the name of substituent before the name of the parent hydrocarbon along with the number of the carbon to which it is attached--Substituents are listed in alphabetical order – neglecting prefixes such as di- tri- tert- etc. -

Deuterium Isotope Effect in the Radiative Triplet Decay of Heavy Atom Substituted Aromatic Molecules
Deuterium Isotope Effect in the Radiative Triplet Decay of Heavy Atom Substituted Aromatic Molecules J. Friedrich, J. Vogel, W. Windhager, and F. Dörr Institut für Physikalische und Theoretische Chemie der Technischen Universität München, D-8000 München 2, Germany (Z. Naturforsch. 31a, 61-70 [1976] ; received December 6, 1975) We studied the effect of deuteration on the radiative decay of the triplet sublevels of naph- thalene and some halogenated derivatives. We found that the influence of deuteration is much more pronounced in the heavy atom substituted than in the parent hydrocarbons. The strongest change upon deuteration is in the radiative decay of the out-of-plane polarized spin state Tx. These findings are consistently related to a second order Herzberg-Teller (HT) spin-orbit coupling. Though we found only a small influence of deuteration on the total radiative rate in naphthalene, a signi- ficantly larger effect is observed in the rate of the 00-transition of the phosphorescence. This result is discussed in terms of a change of the overlap integral of the vibrational groundstates of Tx and S0 upon deuteration. I. Introduction presence of a halogene tends to destroy the selective spin-orbit coupling of the individual triplet sub- Deuterium (d) substitution has proved to be a levels, which is originally present in the parent powerful tool in the investigation of the decay hydrocarbon. If the interpretation via higher order mechanisms of excited states The change in life- HT-coupling is correct, then a heavy atom must time on deuteration provides information on the have a great influence on the radiative d-isotope electronic relaxation processes in large molecules. -

Introduction to Unconventional Superconductivity Manfred Sigrist
Introduction to Unconventional Superconductivity Manfred Sigrist Theoretische Physik, ETH-Hönggerberg, 8093 Zürich, Switzerland Abstract. This lecture gives a basic introduction into some aspects of the unconventionalsupercon- ductivity. First we analyze the conditions to realized unconventional superconductivity in strongly correlated electron systems. Then an introduction of the generalized BCS theory is given and sev- eral key properties of unconventional pairing states are discussed. The phenomenological treatment based on the Ginzburg-Landau formulations provides a view on unconventional superconductivity based on the conceptof symmetry breaking.Finally some aspects of two examples will be discussed: high-temperature superconductivity and spin-triplet superconductivity in Sr2RuO4. Keywords: Unconventional superconductivity, high-temperature superconductivity, Sr2RuO4 INTRODUCTION Superconductivity remains to be one of the most fascinating and intriguing phases of matter even nearly hundred years after its first observation. Owing to the breakthrough in 1957 by Bardeen, Cooper and Schrieffer we understand superconductivity as a conden- sate of electron pairs, so-called Cooper pairs, which form due to an attractive interaction among electrons. In the superconducting materials known until the mid-seventies this interaction is mediated by electron-phonon coupling which gises rise to Cooper pairs in the most symmetric form, i.e. vanishing relative orbital angular momentum and spin sin- glet configuration (nowadays called s-wave pairing). After the introduction of the BCS concept, also studies of alternative pairing forms started. Early on Anderson and Morel [1] as well as Balian and Werthamer [2] investigated superconducting phases which later would be identified as the A- and the B-phase of superfluid 3He [3]. In contrast to the s-wave superconductors the A- and B-phase are characterized by Cooper pairs with an- gular momentum 1 and spin-triplet configuration. -

Marvin Charton - Correlation Analyst Par Excellence
Int. J. Mol. Sci. 2005, 6, 3-10 International Journal of Molecular Sciences ISSN 1422-0067 © 2005 by MDPI www.mdpi.org/ijms/ Marvin Charton - Correlation Analyst Par Excellence John Shorter 29A, Meadowfields, Whitby, North Yorkshire, YO21 1QF, UK. (Emeritus Reader in Chemistry, University of Hull), email: [email protected] Received: 10 September 2004 / Accepted: 19 October 2004 / Published: 31 January 2005 Abstract: Since the late 1950s Marvin Charton, physical organic chemist, of Pratt Institute, Brooklyn, New York has studied structure-property relationships through correlation analysis, and has become one of the leading authorities in this field. Keywords: Hammett equation, correlation analysis, linear free energy relationships, substituent effects and constants, biological activity. Introduction Marvin Charton, of Pratt Institute, Brooklyn, New York, describes his research interests in his CV as follows: "The study of structure-property quantitative relationships in chemistry and biology by means of correlation analysis. Applications of this work include chemical reaction mechanisms, the estimation of chemical and physical properties and chemical reactivities of chemical compounds, the design of bioactive molecules including medicinal drugs and pesticides, and the prediction of environmental toxicities and other properties." His first paper in this field appeared in 1958 and he has now published over 150 research papers and review articles. Marvin's forebears came to the USA from eastern Europe in the late 19th century. His surname may sound French, but his ancestry was Polish, and the relatives who came from Poland to America had the name of Charton. He sometimes suggests, half in fun, that one of his ancestors may have accompanied Napoleon from France to Moscow in 1812, but during the great retreat, by the time they reached Poland he was a little tired of marching, so he absented himself, and found a good place to live and a nice Polish girl to marry... -

And Nitro- Oxy-Polycyclic Aromatic Hydrocarbons
This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organization or the World Health Organization. Environmental Health Criteria 229 SELECTED NITRO- AND NITRO- OXY-POLYCYCLIC AROMATIC HYDROCARBONS First draft prepared by Drs J. Kielhorn, U. Wahnschaffe and I. Mangelsdorf, Fraunhofer Institute of Toxicology and Aerosol Research, Hanover, Germany Please note that the pagination and layout of this web version are not identical to the printed EHC Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organization and the World Health Organization, and produced within the framework of the Inter-Organization Programme for the Sound Management of Chemicals. The International Programme on Chemical Safety (IPCS), established in 1980, is a joint venture of the United Nations Environment Programme (UNEP), the International Labour Organization (ILO) and the World Health Organization (WHO). The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals. The Inter-Organization Programme for the Sound Management of Chemicals (IOMC) was established in 1995 by UNEP, ILO, the Food and Agriculture Organization of the United Nations, WHO, the United Nations Industrial Development Organization, the United Nations Institute for Training and Research and the Organisation for Economic Co- operation and Development (Participating Organizations), following recommendations made by the 1992 UN Conference on Environment and Development to strengthen cooperation and increase coordination in the field of chemical safety. -

II. Nomenclature Rules for Alkenes 1. the Parent Name Will Be the Longest
1 Lecture 9 II. Nomenclature Rules For Alkenes 1. The parent name will be the longest carbon chain that contains both carbons of the double bond. Drop the -ane suffix of the alkane name and add the –ene suffix. Never name the double bond as a prefix. If a double bond is present, you have an alkene, not an alkane. alkane + -ene = alkene 2. Begin numbering the chain at the end nearest the double bond. Always number through the double bond and identify its position in the longest chain with the lower number. In the older IUPAC rules the number for the double bond was placed in front of the stem name with a hyphen. Under the newer rules, the number for the double bond is placed right in front of “ene”, with hyphens. We will use the newer rules for specifying the location of pi bonds. 1 2 3456 H3CCHCH CH 2 CH2 CH3 hex-2-ene (newer rules) 2-hexene (older rules) 3. Indicate the position of any substituent group by the number of the carbon atom in the parent (longest) chain to which it is attached. CH 1 2 345 3 H3CCHCHCHCH2 CH CH3 6 7 CH3 Numbering is determined by the double bond, not the branches, because the double bond has 5,6-dimethylhept-3-ene (newer rules) higher priority than any alkyl branch. 5,6-dimethyl-3-heptene (older rules) 4. Number cycloalkenes so that the double bond is 1,2 (number through the double bond). Number in the direction about the ring so that the lowest number is used at the first point of difference. -
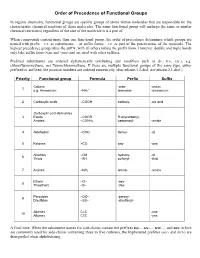
Priority of Functional Groups
Order of Precedence of Functional Groups In organic chemistry, functional groups are specific groups of atoms within molecules that are responsible for the characteristic chemical reactions of those molecules. The same functional group will undergo the same or similar chemical reaction(s) regardless of the size of the molecule it is a part of. When compounds contain more than one functional group, the order of precedence determines which groups are named with prefix – i.e. as substituents –, or suffix forms – i.e. as part of the parent name of the molecule. The highest precedence group takes the suffix, with all others taking the prefix form. However, double and triple bonds only take suffix form (-ene and -yne) and are used with other suffixes. Prefixed substituents are ordered alphabetically (excluding any modifiers such as di-, tri-, etc.), e.g. chlorofluoromethane, not fluorochloromethane. If there are multiple functional groups of the same type, either prefixed or suffixed, the position numbers are ordered numerically (thus ethane-1,2-diol, not ethane-2,1-diol.) Priority Functional group Formula Prefix Suffix Cations -onio- -onium 1 + e.g. Ammonium –NH4 ammonio- -ammonium 2 Carboxylic acids –COOH carboxy- -oic acid Carboxylic acid derivatives 3 Esters –COOR R-oxycarbonyl- Amides –CONH2 carbamoyl- -amide 4 Aldehydes –CHO formyl- -al 5 Ketones >CO oxo- -one Alcohols –OH hydroxy- -ol 6 Thiols –SH sulfanyl- -thiol 7 Amines –NH2 amino- -amine Ethers –O– -oxy- 8 Thioethers –S– -thio- Peroxides –OO– -peroxy- 9 Disulfides –SS– -disulfanyl- Alkenes C=C -ene 10 Alkynes C≡C -yne A final note: When the substituent names for side-chains contain the prefixes iso..., sec-..., tert-..., and neo (which are commonly used for side-chains containing three to five carbons), the hyphenated prefixes (sec- and tert-) are disregarded when alphabetizing. -

PHYSICAL ORGANIC Chemistryl
PHYSICAL ORGANIC CHEMISTRYl By EDWARD R. THORNTON Department of Chemistry, University of Pennsylvania, Philadelphia, Pennsylvania The author, who considers himself to have interests typical of a physical organic chemist, found about 2300 papers published during the last year that were of special interest to him. He confesses that he could not possibly read them all and hopes that those whose work may have been left out by neces sity or oversight will understand the problems involved. Most of this review discusses secondary and solvent isotope effects. Mention is also made of recent developments in quantum chemistry, es pecially in the qualitative discussion of orbital symmetry as a determining factor for certain reaction pathways. It cannot be claimed that enough space was available for comprehensive coverage of even these restricted topics. SECONDARY DEUTERIUM ISOTOPE EFFECTS A great deal of interesting information on secondary isotope effects has become available since the comprehensive review by Halevi (1) in 1963. Advances have been made in both experimental studies and theoretical un derstanding. Some of this new work, along with the present author's inter pretation of secondary isotope effects, will be discussed. Origin of secondary isotope effects.-A great deal of difficulty occurs in attempting to describe the causes of secondary isotope effects. Nevertheless, as far as the author knows, there is no case where an appropriate statistical treatment of isotope effects [Bigeleisen & Wolfsberg (2), Melander (3), Thornton (4)] does not adequately describe experiment. Even in terms of isotopic effects of the order of a few per cent, this theory and the approxima tions it uses are very precise. -

1 Chapter 3: Organic Compounds: Alkanes and Cycloalkanes
Chapter 3: Organic Compounds: Alkanes and Cycloalkanes >11 million organic compounds which are classified into families according to structure and reactivity Functional Group (FG): group of atoms which are part of a large molecule that have characteristic chemical behavior. FG’s behave similarly in every molecule they are part of. The chemistry of the organic molecule is defined by the function groups it contains 1 C C Alkanes Carbon - Carbon Multiple Bonds Carbon-heteroatom single bonds basic C N C C C X X= F, Cl, Br, I amines Alkenes Alkyl Halide H C C C O C C O Alkynes alcohols ethers acidic H H H C S C C C C S C C H sulfides C C thiols (disulfides) H H Arenes Carbonyl-oxygen double bonds (carbonyls) Carbon-nitrogen multiple bonds acidic basic O O O N H C H C O C Cl imine (Schiff base) aldehyde carboxylic acid acid chloride O O O O C C N C C C C O O C C nitrile (cyano group) ketones ester anhydrides O C N amide opsin Lys-NH2 + Lys- opsin H O H N rhodopsin H 2 Alkanes and Alkane Isomers Alkanes: organic compounds with only C-C and C-H single (s) bonds. general formula for alkanes: CnH(2n+2) Saturated hydrocarbons Hydrocarbons: contains only carbon and hydrogen Saturated" contains only single bonds Isomers: compounds with the same chemical formula, but different arrangement of atoms Constitutional isomer: have different connectivities (not limited to alkanes) C H O C4H10 C5H12 2 6 O OH butanol diethyl ether straight-chain or normal hydrocarbons branched hydrocarbons n-butane n-pentane Systematic Nomenclature (IUPAC System) Prefix-Parent-Suffix -

Physical Organic Chemistry
CHM 8304 Physical Organic Chemistry Thermodynamics and kinetics Outline: Isotope effects • see section 8.1 of A&D – experimental approach – primary isotope effect – secondary isotope effect – equilibrium isotope effect – solvent isotope effect – heavy atom isotope effects 2 Thermodynamics and kinetics 1 CHM 8304 Measurement of an isotope effect • performed to determine if a bond changes in a certain way during the rate-limiting step • expressed as a ratio whose numerator is the rate constant measured for the naturally abundant isotope and the denominator is the rate constant measured for the varied isotope – e.g. kH/kD 3 Types of isotope effects • kinetic isotope effects (kie): result from a change in the rate constant of a reaction : – normal effect: ratio > 1 – inverse effect: ratio < 1 – primary isotope effect: when the isotopically substituted bond is cleaved during the rate-limiting step – secondary isotope effect : attributable to a change of hybridation state, not cleavage of bonds • equilibrium isotope effects: result from displacement of an equilibrium 4 Thermodynamics and kinetics 2 CHM 8304 Origin of isotope effects • the origin of all isotope effects is a difference in the frequency of vibrational modes of a substituted molecule with respect to an unsubstituted molecule • it is these vibrational modes that principally affect the shape of the potential energy well on an energy surface 5 Zero point energy • zero point energy (ZPE) is the energy level of the vibrational ground state for most molecules at ambient temperature •