Physical Organic Chemistry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Brief Guide to the Nomenclature of Organic Chemistry
1 Brief Guide to the Nomenclature of Table 1: Components of the substitutive name Organic Chemistry (4S,5E)-4,6-dichlorohept-5-en-2-one for K.-H. Hellwich (Germany), R. M. Hartshorn (New Zealand), CH3 Cl O A. Yerin (Russia), T. Damhus (Denmark), A. T. Hutton (South 4 2 Africa). E-mail: [email protected] Sponsoring body: Cl 6 CH 5 3 IUPAC Division of Chemical Nomenclature and Structure suffix for principal hept(a) parent (heptane) one Representation. characteristic group en(e) unsaturation ending chloro substituent prefix 1 INTRODUCTION di multiplicative prefix S E stereodescriptors CHEMISTRY The universal adoption of an agreed nomenclature is a key tool for 2 4 5 6 locants ( ) enclosing marks efficient communication in the chemical sciences, in industry and Multiplicative prefixes (Table 2) are used when more than one for regulations associated with import/export or health and safety. fragment of a particular kind is present in a structure. Which kind of REPRESENTATION The International Union of Pure and Applied Chemistry (IUPAC) multiplicative prefix is used depends on the complexity of the provides recommendations on many aspects of nomenclature.1 The APPLIED corresponding fragment – e.g. trichloro, but tris(chloromethyl). basics of organic nomenclature are summarized here, and there are companion documents on the nomenclature of inorganic2 and Table 2: Multiplicative prefixes for simple/complicated entities polymer3 chemistry, with hyperlinks to original documents. An No. Simple Complicated No. Simple Complicated AND overall -

Singlet/Triplet State Anti/Aromaticity of Cyclopentadienylcation: Sensitivity to Substituent Effect
Article Singlet/Triplet State Anti/Aromaticity of CyclopentadienylCation: Sensitivity to Substituent Effect Milovan Stojanovi´c 1, Jovana Aleksi´c 1 and Marija Baranac-Stojanovi´c 2,* 1 Institute of Chemistry, Technology and Metallurgy, Center for Chemistry, University of Belgrade, Njegoševa 12, P.O. Box 173, 11000 Belgrade, Serbia; [email protected] (M.S.); [email protected] (J.A.) 2 Faculty of Chemistry, University of Belgrade, Studentski trg 12-16, P.O. Box 158, 11000 Belgrade, Serbia * Correspondence: [email protected]; Tel.: +381-11-3336741 Abstract: It is well known that singlet state aromaticity is quite insensitive to substituent effects, in the case of monosubstitution. In this work, we use density functional theory (DFT) calculations to examine the sensitivity of triplet state aromaticity to substituent effects. For this purpose, we chose the singlet state antiaromatic cyclopentadienyl cation, antiaromaticity of which reverses to triplet state aromaticity, conforming to Baird’s rule. The extent of (anti)aromaticity was evaluated by using structural (HOMA), magnetic (NICS), energetic (ISE), and electronic (EDDBp) criteria. We find that the extent of triplet state aromaticity of monosubstituted cyclopentadienyl cations is weaker than the singlet state aromaticity of benzene and is, thus, slightly more sensitive to substituent effects. As an addition to the existing literature data, we also discuss substituent effects on singlet state antiaromaticity of cyclopentadienyl cation. Citation: Stojanovi´c,M.; Aleksi´c,J.; Baranac-Stojanovi´c,M. Keywords: antiaromaticity; aromaticity; singlet state; triplet state; cyclopentadienyl cation; substituent Singlet/Triplet State effect Anti/Aromaticity of CyclopentadienylCation: Sensitivity to Substituent Effect. -

Inductive Effect
Dr Anju K. Gupta Associate Professor Department of Chemistry A.N. College, Patna Lecture note for B.Sc. (I) Subsidiary Inductive Effect If a covalent bond is formed between atoms having similar electronegativity, the bonding electrons will be shared equally between the two atoms. For example, in hydrogen molecule, the two hydrogen atoms have identical electronegativities. Therefore, the electron pair is shared equally between the two hydrogen atoms and so the molecular orbital (σ-bond) will be symmetrically distributed. Similarly, if we consider a chlorine molecule, the molecular orbital (σ- bond) formed between the two identical chlorine atoms is symmetrically distributed. This type of bond is called a non-polar covalent bond. However, if we consider a hydrogen chloride molecule, the electron pair is not equally shared between hydrogen and chlorine because chlorine has a greater electronegativity than hydrogen. Therefore, the electron pair shifts towards the more electronegative atom. This causes polarization in the bond in which the more electronegative atom chlorine acquires a partial positive charge. This type of bond in which unequal sharing of electron pair between two atoms of different electronegativities are involved is called a polar covalent bond. The atom which has a greater share of the paired electrons is given a symbol δ- and the atom with a smaller share is given a symbol δ+. Thus, a polar covalent bond can be represented as: 1 Dr Anju K. Gupta Associate Professor Department of Chemistry A.N. College, Patna Lecture note for B.Sc. (I) Subsidiary Consider a chain of carbon atoms such as C4—C3—C2—C1—X with an atom X at the terminal having higher electronegativity than carbon. -

Stabilizing a Different Cyclooctatetraene Stereoisomer
Stabilizing a different cyclooctatetraene stereoisomer Longfei Lia,b, Ming Leia,1, Yaoming Xieb, Henry F. Schaefer IIIb,1, Bo Chenc, and Roald Hoffmannc,1 aState Key Laboratory of Chemical Resource Engineering, Institute of Materia Medica, College of Science, Beijing University of Chemical Technology, Beijing 100029, People’s Republic of China; bCenter for Computational Quantum Chemistry, University of Georgia, Athens, GA 30602; and cDepartment of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14853-1301 Contributed by Roald Hoffmann, July 27, 2017 (sent for review June 6, 2017; reviewed by Henry Rzepa and Jay S. Siegel) An unconventional cis-cis-cis-trans or (Z,Z,Z,E) structure B of cyclo- (Z,Z,Z,E)-COT octatetraene (COT) is calculated to lie only 23 kcal/mol above the A Second COT Isomer. As did the above-cited authors, we located the well-known tub-shaped (Z,Z,Z,Z) isomer A; one example of this type (Z,Z,Z,E) isomer of COT (B) as a local minimum on its potential of structure is known. The barrier for B returning to A is small, energy surface. Throughout this paper, we use the ωB97X-D/cc- 3 kcal/mol. However, by suitable choice of substituents, the (Z,Z,Z,E) pVTZ (density functional theory method using the ωb97X-D func- isomer can be made to lie in energy below the tub-shaped structure. tional with correlation-consistent polarized valence triple-zeta basis Steric, clamping, and electronic strategies are proposed for achieving set) method of calculation (details are provided in SI Appendix) t B this. -

The Case of Sterimol Steric Parameters A
Conformational Effects on Physical-Organic Descriptors – the Case of Sterimol Steric Parameters A. V. Brethomé,† S. P. Fletcher†* and R. S. Paton†§* †Chemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford OX1 3TA, UK §Department of Chemistry, Colorado State University, Fort Collins, Colorado 80523, USA Abstract Mathematical relationships which relate chemical structure with selectivity have provided quantitative insights underlying catalyst design and informing mechanistic studies. Flexible compounds, however, can adopt several distinct geometries and so can be challenging to describe using a single structure-based descriptor. How best to quantify the structural characteristics of an ensemble of structure poses both practical and technical difficulties. In this work we introduce an automated computational workflow which can be used to obtain multidimensional Sterimol parameters for a conformational ensemble of a given substituent from a single command. The Boltzmann-weighted Sterimol parameters obtained from this approach are shown to be useful in multivariate models of enantioselectivity, while the range of values from conformers within 3 kcal/mol of the most stable structure provides a visual way to capture a possible source of uncertainty arising in the resulting models. Implementing our approach requires no programming expertise and can be executed from within a graphical user interface using open-source programs. Introduction Steric effects are key nonbonding interactions influencing molecular conformation and -

Polar Covalent Bonds: Electronegativity
Polar Covalent Bonds: Electronegativity Covalent bonds can have ionic character These are polar covalent bonds Bonding electrons attracted more strongly by one atom than by the other Electron distribution between atoms is not symmetrical Bond Polarity and Electronegativity Symmetrical Covalent Bonds Polar Covalent Bonds C – C + - C – H C – O (non-polar) (polar) Electronegativity (EN): intrinsic ability of an atom to attract the shared electrons in a covalent bond Inductive Effect: shifting of sigma bonded electrons in resppygonse to nearby electronegative atom The Periodic Table and Electronegativity C – H C - Br and C - I (non-pol)lar) (po l)lar) Bond Polarity and Inductive Effect Nonpolar Covalent Bonds: atoms with similar EN Polar Covalent Bonds: Difference in EN of atoms < 2 Ionic Bonds: Difference in EN > 2 C–H bonds, relatively nonpolar C-O, C-X bonds (more electronegative elements) are polar Bonding electrons shift toward electronegative atom C acqqppuires partial positive char g,ge, + Electronegative atom acquires partial negative charge, - Inductive effect: shifting of electrons in a bond in response to EN of nearby atoms Electrostatic Potential Maps Electrostatic potential maps show calculated charge distributions Colors indicate electron- rich (red) and electron- poor (blue ) reg ions Arrows indicate direction of bond polarity Polar Covalent Bonds: Net Dipole Moments Molecules as a whole are often polar from vector summation of individual bond polarities and lone-pair contributions Strongly polar substances soluble in polar solvents like water; nonpolar substances are insoluble in water. Dipole moment ( ) - Net molecular polarity, due to difference in summed charges - magnitude of charge Q at end of molecular dipole times distance r between charges = Q r, in debyes (D), 1 D = 3.336 1030 coulomb meter length of an average covalent bond, the dipole moment would be 1.60 1029 Cm, or 4.80 D. -

Topological Model on the Inductive Effect in Alkyl Halides Using Local Quantum Similarity and Reactivity Descriptors in the Density Functional Theory
Hindawi Publishing Corporation Journal of Quantum Chemistry Volume 2014, Article ID 850163, 12 pages http://dx.doi.org/10.1155/2014/850163 Research Article Topological Model on the Inductive Effect in Alkyl Halides Using Local Quantum Similarity and Reactivity Descriptors in the Density Functional Theory Alejandro Morales-Bayuelo1,2 and Ricardo Vivas-Reyes1 1 Grupo de Qu´ımica Cuantica´ y Teorica,´ Universidad de Cartagena, Programa de Qu´ımica, Facultad de Ciencias Exactas y Naturales, Cartagena de Indias, Colombia 2 Departamento de Ciencias Qu´ımicas, Universidad Nacional Andres Bello, Republica 275, Santiago, Chile Correspondence should be addressed to Alejandro Morales-Bayuelo; [email protected] and Ricardo Vivas-Reyes; [email protected] Received 19 September 2013; Revised 16 December 2013; Accepted 16 December 2013; Published 19 February 2014 Academic Editor: Daniel Glossman-Mitnik Copyright © 2014 A. Morales-Bayuelo and R. Vivas-Reyes. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. We present a topological analysis to the inductive effect through steric and electrostatic scales of quantitative convergence. Using the molecular similarity field based in the local guantum similarity (LQS) with the Topo-Geometrical Superposition Algorithm (TGSA) alignment method and the chemical reactivity in the density function theory (DFT) context, all calculations were carried out with Amsterdam Density Functional (ADF) code, using the gradient generalized approximation (GGA) and local exchange correlations PW91, in order to characterize the electronic effect by atomic size in the halogens group using a standard Slater-type- orbital basis set. -

Nomenclature of Alkanes
Nomenclature of alkanes methane CH4 ethane CH3CH3 propane CH3CH2CH3 butane CH3CH2CH2CH3 pentane CH3CH2CH2CH2CH3 hexane CH3CH2CH2CH2CH2CH3 heptane CH3CH2CH2CH2CH2CH2CH3 octane CH3CH2CH2CH2CH2CH2CH2CH3 nonane CH3CH2CH2CH2CH2CH2CH2CH2CH3 decane CH3CH2CH2CH2CH2CH2CH2CH2CH2CH3 undecane CH3CH2CH2CH2CH2CH2CH2CH2CH2CH2CH3 dodecane CH3CH2CH2CH2CH2CH2CH2CH2CH2CH2CH2CH3 Funky groups/alkanes iso- CH3 R isopropyl HC R CH3 isobutane/isobutyl R = CH3/CH2R (4 C’s) R isopentane/isopentyl R = CH2CH3/CH2CH2R (5 C’s) R isohexane/isohexyl R = CH2CH2CH3/CH2CH2R (6 C’s) R neo- CH3 neopentyl H3C C CH2 R R CH3 neopentane R = H (5 C’s) neohaxane/neohexyl R = CH3/CH2R (6 C’s) R 1° primary (n-) 2° secondary (sec-, s-) 3° tertiary (tert-, t-) H C H C H3C 3 3 CH CH2 CH CH2 CH CH2 H C CH H C CH H3C CH3 3 3 3 3 n- often used with strait chain compounds though it is not actually necessary. sec- sec-butyl H the substituent is attached s-butyl to a 2° C of butane (4 C’s) H3C CH2 C R CH3 no other "sec-" group tert- tert-butyl CH3 a substituent is attached to t-butyl the 3° C of a 4 C H3C C R molecule/unit CH3 tert-pentyl CH3 a substituent is attached to t-pentyl the 3° of a 5 C molecule/unit H3C CH2 C R CH3 no other "tert-" group Form of name #-followed by substituent name followed by parent hydrocarbon name • Determine longest continuous chain. o This is the parent hydrocarbon o If compound has two or more chains of the same length, parent hydrocarbon is chain with greatest number of substituents • Cite the name of substituent before the name of the parent hydrocarbon along with the number of the carbon to which it is attached--Substituents are listed in alphabetical order – neglecting prefixes such as di- tri- tert- etc. -

Heterocycles 2 Daniel Palleros
Heterocycles 2 Daniel Palleros Heterocycles 1. Structures 2. Aromaticity and Basicity 2.1 Pyrrole 2.2 Imidazole 2.3 Pyridine 2.4 Pyrimidine 2.5 Purine 3. Π-excessive and Π-deficient Heterocycles 4. Electrophilic Aromatic Substitution 5. Oxidation-Reduction 6. DNA and RNA Bases 7. Tautomers 8. H-bond Formation 9. Absorption of UV Radiation 10. Reactions and Mutations Heterocycles 3 Daniel Palleros Heterocycles Heterocycles are cyclic compounds in which one or more atoms of the ring are heteroatoms: O, N, S, P, etc. They are present in many biologically important molecules such as amino acids, nucleic acids and hormones. They are also indispensable components of pharmaceuticals and therapeutic drugs. Caffeine, sildenafil (the active ingredient in Viagra), acyclovir (an antiviral agent), clopidogrel (an antiplatelet agent) and nicotine, they all have heterocyclic systems. O CH3 N HN O O N O CH 3 N H3C N N HN N OH O S O H N N N 2 N O N N O CH3 N CH3 caffeine sildenafil acyclovir Cl S N CH3 N N H COOCH3 nicotine (S)-clopidogrel Here we will discuss the chemistry of this important group of compounds beginning with the simplest rings and continuing to more complex systems such as those present in nucleic acids. Heterocycles 4 Daniel Palleros 1. Structures Some of the most important heterocycles are shown below. Note that they have five or six-membered rings such as pyrrole and pyridine or polycyclic ring systems such as quinoline and purine. Imidazole, pyrimidine and purine play a very important role in the chemistry of nucleic acids and are highlighted. -

Steric Effects Vs. Electron Delocalization
RSC Advances PAPER View Article Online View Journal | View Issue Steric effects vs. electron delocalization: a new look into the stability of diastereomers, conformers Cite this: RSC Adv.,2021,11, 20691 and constitutional isomers† Sopanant Datta and Taweetham Limpanuparb * A quantum chemical investigation of the stability of compounds with identical formulas was carried out on 23 classes of compounds made of C, N, P, O and S atoms as core structures and halogens H, F, Cl, Br and I as Received 13th April 2021 substituents. All possible structures were generated and investigated by quantum mechanical methods. The Accepted 24th May 2021 prevalence of a formula in which its Z configuration, gauche conformation or meta isomer is the most stable DOI: 10.1039/d1ra02877d form is calculated and discussed. Quantitative and qualitative models to explain the stability of the 23 classes rsc.li/rsc-advances of halogenated compounds were also proposed. hyperconjugation in a similar vein to the ethane case,17,18 but Creative Commons Attribution-NonCommercial 3.0 Unported Licence. 1. Introduction the reasoning underlying the preference for meta isomer is still Steric effects, non-bonded interactions leading to avoidance of lacking. spatial congestion of atoms or groups, are oen the central In addition to carbon-backbone compounds in Table 1, there theme in the discussion of stability of diastereomers, are many experimental and theoretical studies for other back- 21 ] 22,23 ] 24 conformers and constitutional isomers. Reasoning based on bones discussed in this paper, namely, C3, C N, C P, 25–31 32 33,34 35 36,37 38–40 41 steric effects is relatively intuitive and gives rise to a generally N]N, N]P, P]P, C–N, C–P, C–O, C–S, N– 38,42–44 38,43 38,43,45,46 38,42,47 47 48,49 50 accepted rule of thumb that an E conguration, anti conformer N, N–P, P–P, N–O, N–S, P–O, P–S, O– 38,42,51,52 53,54 38,55 and para isomer in diastereomers, conformational and consti- O, O–S, and S–S. -
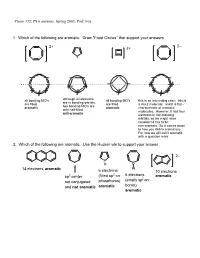
PS 6 Answers, Spring 2003, Prof
Chem 332, PS 6 answers, Spring 2003, Prof. Fox 1. Which of the following are aromatic. Draw "Frost Circles" that support your answers 2– 2+ 2+ + all bonding MO's although all electrons all bonding MO's this is an interesting case: this is are in bonding orbitals, are filled are filled a 4n+2 molecule, and it is flat-- two bonding MO's are aromatic aromatic characteristic of aromatic only half-filled molecules. However, it has four anti-aromatic electrons in non-bonding orbitals, so we might have considered this to be non-aromatic. So it comes down to how you define aromaticity. For now we will call it aromatic with a question mark. 2. Which of the following are aromatic. Use the Huckel rule to support your answer 2– P H B 14 electrons, aromatic H 6 electrons 10 electrons 2 6 electrons sp3 center (filled sp on aromatic (empty sp2 on not conjugated phosphorus) boron) and not aromatic aromatic aromatic 3. Compound 1 has an unusually large dipole moment for an organic hydrocarbon. Explain why (consider resonance structures for the central double bond) We need to remember that for any double bond compound, we can draw two polar resonance structure. Normally, these resonance structures are unimportant unimportant unimportant However, for 1 the story is different because there is a polar resonance form that has two aromatic components. In other words, 'breaking' the double bond gives rise to aromaticity, and therefore the polar resonance structure is important and leads to the observed dipole. dipole 1 8 e– 4 e– 6 e– 6 e– anti- anti- aromatic aromatic aromatic aromatic important resonance unimportant resonance structure structure 5. -

Structure of Benzene, Orbital Picture, Resonance in Benzene, Aromatic Characters, Huckel’S Rule B
Dr.Mrityunjay Banerjee, IPT, Salipur B PHARM 3rd SEMESTER BP301T PHARMACEUTICAL ORGANIC CHEMISTRY –II UNIT I UNIT I Benzene and its derivatives 10 Hours A. Analytical, synthetic and other evidences in the derivation of structure of benzene, Orbital picture, resonance in benzene, aromatic characters, Huckel’s rule B. Reactions of benzene - nitration, sulphonation, halogenationreactivity, Friedelcrafts alkylation- reactivity, limitations, Friedelcrafts acylation. C. Substituents, effect of substituents on reactivity and orientation of mono substituted benzene compounds towards electrophilic substitution reaction D. Structure and uses of DDT, Saccharin, BHC and Chloramine Benzene and its Derivatives Chemists have found it useful to divide all organic compounds into two broad classes: aliphatic compounds and aromatic compounds. The original meanings of the words "aliphatic" (fatty) and "aromatic" (fragrant/ pleasant smell). Aromatic compounds are benzene and compounds that resemble benzene in chemical behavior. Aromatic properties are those properties of benzene that distinguish it from aliphatic hydrocarbons. Benzene: A liquid that smells like gasoline Boils at 80°C & Freezes at 5.5°C It was formerly used to decaffeinate coffee and component of many consumer products, such as paint strippers, rubber cements, and home dry-cleaning spot removers. A precursor in the production of plastics (such as Styrofoam and nylon), drugs, detergents, synthetic rubber, pesticides, and dyes. It is used as a solvent in cleaning and maintaining printing equipment and for adhesives such as those used to attach soles to shoes. Benzene is a natural constituent of petroleum products, but because it is a known carcinogen, its use as an additive in gasoline is now limited. In 1970s it was associated with leukemia deaths.