Physiology of the Lung in Idiopathic Pulmonary Fibrosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lung Function Studies in Diagnostics and Follow-Up of Pulmonary Sarcoidosis
Lung Function Studies in Diagnostics and Follow-up of Pulmonary Sarcoidosis By INGELA BRÅDVIK Lund 1994 From the Department of Lung Medicine and the Department of Clinical Physiology University of Lund, Sweden Lung Function Studies in Diagnostics and Follow-up of Pulmonary Sarcoidosis Ingela Brådvik Lund 1994 Organization Document name LUND UNIVERSITY DOCTORAL DISSERTATION Department of Lung Medicine Date of issue University Hospital 94 06 09 S-22I 85 Lund CODEN: ISRN LUMEDW/MELL- - 1007- - SE Authorfj) Sponsoring organization Ingela Brådvik The Swedish Heart Lung Foundation Title and subtitle Lung function studies in diagnostics and follow-up of pulmonary sarcoidosis Abstract In 66 patients the relationship between lung volumes and lung mechanics in pulmonary sarcoidosis was investigated Lung volumes, static lung mechanics, lung resistance, dynamic lung mechanics and arterial blood gases at rest and during exercise were obtained. Fifteen functionally compromised patients received steroids during one year. They were re-investigated during the treatment and at a follow-up after an average of 7 years. In another 41 patients with newly diagnosed sarcoidosis, the kinetics of the lung clearance of ^9mTc-DTPA measured over 180 minutes was explored, and compared to kinetics in healthy smokers. The relationship between lung clearance and lung volumes, lung mechanics, arterial blood gases and disease activity assessed with serum angiotensin-converting enzyme and "'Ga scintigraphy was studied. Reduced lung volumes and compliance, increased resistance and decreased arterial oxygen tension were common. Vital i capacity (VC), and changes of VC at follow-up, corresponded to the slope of the static elastic pressure/volume curve, . and to the variation of it. -

Bronchodilators Accelerate the Dynamics of Muscle O2 Delivery
Chronic obstructive pulmonary disease Bronchodilators accelerate the dynamics of muscle Thorax: first published as 10.1136/thx.2009.120857 on 13 July 2010. Downloaded from O2 delivery and utilisation during exercise in COPD Danilo C Berton,1 Priscila B Barbosa,1 Luciana S Takara,1 Gaspar R Chiappa,1 Ana Cristina B Siqueira,1 Daniela M Bravo,1 Leonardo F Ferreira,2 J Alberto Neder1 See Editorial, p 573 ABSTRACT (QT), suggesting an important role for the central Background Expiratory flow limitation and lung cardiovascular adjustments in setting the limits of < Supplementary methods are hyperinflation promote cardiocirculatory perturbations increase in peripheral QO at the onset of exercise. published online only. To view 2 these files please visit the that might impair O2 delivery to locomotor muscles in Further experimental evidence for this contention journal online (http://thorax.bmj. patients with chronic obstructive pulmonary disease was obtained in a subsequent study in which com). (COPD). The hypothesis that decreases in lung hyperinflation a strategy aimed to reduce the resistive work of 1Pulmonary Function and Clinical after the inhalation of bronchodilators would improve breathing and dynamic hyperinflation (heliox) Exercise Physiology Unit skeletal muscle oxygenation during exercise was tested. simultaneously accelerated the dynamics of QTand (SEFICE), Division of Respiratory 5 Methods Twelve non- or mildly hypoxaemic males QO2 in patients with moderate to severe COPD. Diseases, Department of (forced expiratory volume in 1 s (FEV1)¼38.5612.9% -

Coding Billing
CodingCoding&Billing FEBRUARY 2020 Quarterly Editor’s Letter Welcome to the February issue of the ATS Coding and Billing Quarterly. There are several important updates about the final Medicare rules for 2020 that will be important for pulmonary, critical care and sleep providers. Additionally, there is discussion of E/M documentation rules that will be coming in 2021 that practices might need some time to prepare for, and as always, we will answer coding, billing and regulatory compliance questions submitted from ATS members. If you are looking for a more interactive way to learn about the 2020 Medicare final rules, there is a webinar on the ATS website that covers key parts of the Medicare final rules. But before we get to all this important information, I have a request for your help. EDITOR ATS Needs Your Help – Recent Invoices for Bronchoscopes and PFT Lab ALAN L. PLUMMER, MD Spirometers ATS RUC Advisor TheA TS is looking for invoices for recently purchased bronchoscopes and ADVISORY BOARD MEMBERS: PFT lab spirometer. These invoices will be used by theA TS to present practice KEVIN KOVITZ, MD expense cost equipment to CMS to help establish appropriate reimbursement Chair, ATS Clinical Practice Committee rates for physician services using this equipment. KATINA NICOLACAKIS, MD Member, ATS Clinical Practice Committee • Invoices should not include education or service contract as those ATS Alternate RUC Advisorr are overhead and cannot be considered by CMS for this portion of the STEPHEN P. HOFFMANN, MD Member, ATS Clinical Practice Committee formula and payment rates. ATS CPT Advisor • Invoices can be up to five years old. -

The Pulmonary Manifestations of Left Heart Failure*
The Pulmonary Manifestations of Left Heart Failure* Brian K. Gehlbach, MD; and Eugene Geppert, MD Determining whether a patient’s symptoms are the result of heart or lung disease requires an understanding of the influence of pulmonary venous hypertension on lung function. Herein, we describe the effects of acute and chronic elevations of pulmonary venous pressure on the mechanical and gas-exchanging properties of the lung. The mechanisms responsible for various symptoms of congestive heart failure are described, and the significance of sleep-disordered breathing in patients with heart disease is considered. While the initial clinical evaluation of patients with dyspnea is imprecise, measurement of B-type natriuretic peptide levels may prove useful in this setting. (CHEST 2004; 125:669–682) Key words: Cheyne-Stokes respiration; congestive heart failure; differential diagnosis; dyspnea; pulmonary edema; respiratory function tests; sleep apnea syndromes Abbreviations: CHF ϭ congestive heart failure; CSR-CSA ϭ Cheyne-Stokes respiration with central sleep apnea; CPAP ϭ continuous positive airway pressure; Dlco ϭ diffusing capacity of the lung for carbon monoxide; DM ϭ membrane conductance; FRC ϭ functional residual capacity; OSA ϭ obstructive sleep apnea; TLC ϭ total lung ϭ ˙ ˙ ϭ capacity; VC capillary volume; Ve/Vco2 ventilatory equivalent for carbon dioxide early 5 million Americans have congestive heart For a detailed review of the pathophysiology of N failure (CHF), with 400,000 new cases diag- high-pressure pulmonary edema, the reader is re- nosed each year.1 Unfortunately, despite the consid- ferred to several excellent recent reviews.2–4 erable progress that has been made in understanding the pathophysiology of pulmonary edema, the pul- monary complications of this condition continue to The Pathophysiology of Pulmonary challenge the bedside clinician. -

Effect of Heliox Breathing on Flow Limitation in Chronic Heart Failure Patients
Eur Respir J 2009; 33: 1367–1373 DOI: 10.1183/09031936.00117508 CopyrightßERS Journals Ltd 2009 Effect of heliox breathing on flow limitation in chronic heart failure patients M. Pecchiari*, T. Anagnostakos#, E. D’Angelo*, C. Roussos#, S. Nanas# and A. Koutsoukou# ABSTRACT: Patients with chronic heart failure (CHF) exhibit orthopnoea and tidal expiratory flow AFFILIATIONS limitation in the supine position. It is not known whether the flow-limiting segment occurs in the *Istituto di Fisiologia Umana I, Universita` degli Studi di Milano, peripheral or central part of the tracheobronchial tree. The location of the flow-limiting segment Milan, Italy, and can be inferred from the effects of heliox (80% helium/20% oxygen) administration. If maximal #Dept of Critical Care and Pulmonary expiratory flow increases with this low-density mixture, the choke point should be located in the Services, Evangelismos General central airways, where the wave-speed mechanism dominates. If the choke point were located in Hospital, Medical School, University of Athens, Athens, Greece. the peripheral airways, where maximal flow is limited by a viscous mechanism, heliox should have no effect on flow limitation and dynamic hyperinflation. CORRESPONDENCE Tidal expiratory flow limitation, dynamic hyperinflation and breathing pattern were assessed in M. Pecchiari 14 stable CHF patients during air and heliox breathing at rest in the sitting and supine position. Istituto di Fisiologia Umana I via L. Mangiagalli 32 No patient was flow-limited in the sitting position. In the supine posture, eight patients exhibited 20133 Milan tidal expiratory flow limitation on air. Heliox had no effect on flow limitation and dynamic Italy hyperinflation and only minor effects on the breathing pattern. -

Vocal Cord Dysfunction: Analysis of 27 Cases and Updated Review of Pathophysiology & Management
THIEME Original Research 125 Vocal Cord Dysfunction: Analysis of 27 Cases and Updated Review of Pathophysiology & Management Shibu George1 Sandeep Suresh2 1 Department of ENT, Government Medical College, Kottayam, Kerala, India Address for correspondence ShibuGeorge,MS,DNB,Charivukalayil 2 Department of ENT, Little Flower Hospital, Ernakulam, Kerala, India (House), Ettumanoor, Kottayam 686631, Kerala, India (e-mail: [email protected]; [email protected]). Int Arch Otorhinolaryngol 2019;23:125–130. Abstract Introduction Vocal cord dysfunction is characterized by unintentional paradoxical vocal cord movement resulting in abnormal inappropriate adduction, especially during inspiration; this predominantly manifests as unresponsive asthma or unexplained stridor. It is prudent to be well informed about the condition, since the primary presentation may mask other airway disorders. Objective This descriptive study was intended to analyze presentations of vocal cord dysfunction in a tertiary care referral hospital. The current understanding regarding the pathophysiology and management of the condition were also explored. Methods A total of 27 patients diagnosed with vocal cord dysfunction were analyzed based on demographic characteristics, presentations, associations and examination findings. The mechanism of causation, etiological factors implicated, diagnostic considerations and treatment options were evaluated by analysis of the current literature. Results Therewasastrongfemalepredilection noted among the study population (n ¼ 27), which had a mean age of 31. The most common presentations were stridor Keywords (44%) and refractory asthma (41%). Laryngopharyngeal reflux disease was the most ► Vocal Cord common association in the majority (66%) of the patients, with a strong overlay of Dysfunction anxiety, demonstrable in 48% of the patients. ► paradoxical vocal Conclusion Being aware of the condition is key to avoid misdiagnosis in vocal cord cord motion dysfunction. -

Interstitial Lung Disease—Raising the Index of Suspicion in Primary Care
www.nature.com/npjpcrm All rights reserved 2055-1010/14 PERSPECTIVE OPEN Interstitial lung disease: raising the index of suspicion in primary care Joseph D Zibrak1 and David Price2 Interstitial lung disease (ILD) describes a group of diseases that cause progressive scarring of the lung tissue through inflammation and fibrosis. The most common form of ILD is idiopathic pulmonary fibrosis, which has a poor prognosis. ILD is rare and mainly a disease of the middle-aged and elderly. The symptoms of ILD—chronic dyspnoea and cough—are easily confused with the symptoms of more common diseases, particularly chronic obstructive pulmonary disease and heart failure. ILD is infrequently seen in primary care and a precise diagnosis of these disorders can be challenging for physicians who rarely encounter them. Confirming a diagnosis of ILD requires specialist expertise and review of a high-resolution computed tomography scan (HRCT). Primary care physicians (PCPs) play a key role in facilitating the diagnosis of ILD by referring patients with concerning symptoms to a pulmonologist and, in some cases, by ordering HRCTs. In our article, we highlight the importance of prompt diagnosis of ILD and describe the circumstances in which a PCP’s suspicion for ILD should be raised in a patient presenting with chronic dyspnoea on exertion, once more common causes of dyspnoea have been investigated and excluded. npj Primary Care Respiratory Medicine (2014) 24, 14054; doi:10.1038/npjpcrm.2014.54; published online 11 September 2014 INTRODUCTION emphysema, in which the airways of the lungs become narrowed Interstitial lung disease (ILD) is an umbrella term, synonymous or blocked so the patient cannot exhale completely. -

The Measurement of Pulmonary Diffusing Capacity for Carbon Monoxide by a Rebreathing Method
THE MEASUREMENT OF PULMONARY DIFFUSING CAPACITY FOR CARBON MONOXIDE BY A REBREATHING METHOD Benjamin M. Lewis, … , Ernest J. Hayford-Welsing, Erma Flaherty J Clin Invest. 1959;38(11):2073-2086. https://doi.org/10.1172/JCI103985. Research Article Find the latest version: https://jci.me/103985/pdf THE MEASUREMENT OF PULMONARY DIFFUSING CAPACITY FOR CARBON MONOXIDE BY A REBREATHING METHOD*t By BENJAMIN M. LEWIS, TAI-HON LIN, FRANCES E. NOET AND ERNEST J. HAYFORD-WELSING§ WITH THE TECHNICAL ASSISTANCE OF ERMA FLAHERTY (From the Pulmonary Function Laboratories, Departments of Medicine, Wayne State University College of Medicine, and City of Detroit Receiving Hospital, Detroit, Mich.) (Submitted for publication February 18, 1959; accepted June 19, 1959) The pulmonary diffusing capacity for oxygen capacity and one second vital capacity are first determined. is of great physiological and clinical sig- The analyzer circuit of the apparatus is then flushed with (DLo,) tank oxygen. A sealed bag containing a volume of 0.3 nificance (1). Its measurement, however, is rela- per cent CO and 10 per cent He in air (or in oxygen)2 tively complex (2). Pulmonary diffusing capac- equal to the subject's one second vital capacity is attached ity for carbon monoxide (DLco) which, it is to the three-way tap, the clamps on the bag are removed usually assumed,' can be converted to DL02 from and the bag and analyzer circuit mixed by the pump.3 the known solubilities and molecular weights of A bag-in-box device attached to a spirometer has been found convenient for filling the bag. -

Making the Diagnosis of Asthma
Making the Diagnosis of Asthma Meredith C McCormack MD MHS and Paul L Enright MD Introduction Signs and Symptoms Differential Diagnosis Diagnostic Testing Spirometry Bronchodilator Response Testing Inhalation Challenge Test Radioallergosorbent Test and Allergen Skin Test Exhaled Nitric Oxide Radiographic Imaging Asthma Versus Chronic Obstructive Pulmonary Disease Asthma Severity Summary: Take-Home Messages for the Respiratory Therapist Diagnostic tests can only increase or decrease the probability of the asthma diagnosis, so a thorough history is very important. In patients with asthma-like symptoms, spirometric evidence of airway obstruction plus a large bronchodilator response makes asthma much more likely. However, nor- mal spirometry is common in patients with mild asthma who are not symptomatic at the time of testing, and patients with poorly controlled asthma may lack substantial bronchodilator response. Inhalation challenge test often helps confirm asthma in patients with normal spirometry. Adult smokers with intermittent respiratory symptoms may have either asthma or chronic obstructive pulmonary disease (COPD). Normal post-bronchodilator spirometry rules out COPD. In patients with airway obstruction, a low diffusing capacity of the lung for carbon monoxide increases the probability of COPD and makes asthma much less likely. A high exhaled nitric oxide level makes allergic asthma more likely. Response to inhaled corticosteroids makes asthma more likely and COPD less likely. Key words: asthma, spirometry, methacholine, bronchodilator, pulmonary function, diagnosis. [Respir Care 2008;53(5):583–590. © 2008 Daedalus Enterprises] Meredith C McCormack MD MHS is affiliated with the Division of consulting on spirometry quality-assurance programs for a phase-3 clin- Pulmonary and Critical Care Medicine, Johns Hopkins School of Med- ical trial of varenicline for smoking cessation. -

Submitting Requests for Prior Authorization
Molina Healthcare/Molina Medicare of California Prior Authorization/Pre-Service Review Guide Effective: 08/01/2012 This Prior Authorization/Pre-Service Guide applies to all Molina Healthcare/Molina Medicare Members /Sacrament LIHP. ***Referrals to Network Specialists do not require Prior Authorization*** Authorization required for services listed below. Pre-Service Review is required for elective services. Only covered services will be paid. If you are contracted with Molina through an IPA / Medical Group please refer to your IPA / MG Prior Authorization requirements. For San Diego Medi-Cal members age 0 – 17.99 years old please refer to Children’s Physicians Medical Group’s (CPMG) Prior Authorization requirements. All Non-Par providers/services: services, including office Hearing Aids – including bone anchored hearing aids. visits, provided by non-participating providers, facilities and Not a covered benefit for Sacramento LIHP labs, except professional services related to ER visit, approved Home Healthcare: after initial 3 skilled nursing Ambulatory Surgical Center or inpatient stay and Women’s visits health/OB services. ER visits do not require PA Home Infusion Alcohol and Chemical Dependency Services (Medicare & Outpatient Hospice & Palliative Care: notification CHIP only) Refer to Comp Care or Behavioral Health contact information – page 3 only. All Inpatient Admissions: Acute hospital, SNF, Rehab, Not a covered benefit for Sacramento LIHP LTACS, Hospice(notification only) Behavioral Health Services: - Inpatient, Partial Imaging: CT, MRI, MRA, PET, SPECT, Cardiac Nuclear hospitalization, Day Treatment, Intensive Outpatient Programs Studies, CT Angiograms, intimal media thickness (IOP), ECT, and > 12 Office Visits/year for adults and 20 Office testing, three dimensional imaging visits/year for children (Medicare & CHIP only) Refer to Behavioral Neuropsychological Testing and Therapy. -

Middle Airway Obstructiondit May Be Happening Under Our Noses Philip G Bardin,1 Sebastian L Johnston,2 Garun Hamilton1
Thorax Online First, published on July 19, 2012 as 10.1136/thoraxjnl-2012-202221 Chest clinic OPINION Thorax: first published as 10.1136/thoraxjnl-2012-202221 on 19 July 2012. Downloaded from Middle airway obstructiondit may be happening under our noses Philip G Bardin,1 Sebastian L Johnston,2 Garun Hamilton1 < Additional materials are ABSTRACT for this conceptual error to be refuted.2 The virus is published online only. To view Background Lower airway obstruction has evolved to now understood to spread from nose to lung, and these files please visit the denote pathologies associated with diseases of the lung, appropriate recognition of the close association journal online (http://dx.doi.org/ 10.1136/ whereas, conditions proximal to the lung embody upper between upper and lower airway pathologies has thoraxjnl-2012-202221/content/ airway obstruction. This approach has disconnected had positive outcomes. An example is novel strat- early/recent). diseases of the larynx and trachea from the lung, and egies that may not be able to prevent colds, but 1Lung and Sleep Medicine, removed the ‘middle airway’ from the interest and that could ameliorate virus asthma exacerbations Monash University and Hospital involvement of respiratory physicians and scientists. through the use of inhaled interferon.3 and Monash Institute of Medical However, recent studies have indicated that dysfunction Accumulating evidence suggests that the middle Research (MIMR), Melbourne, of this anatomical region may be a key component of Australia airway plays a key role in airway obstruction either 2Respiratory Medicine, Imperial overall airway obstruction, either independently or in independently or with coexisting lung disease. -
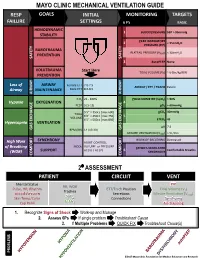
Mechanical Ventilation Guide
MAYO CLINIC MECHANICAL VENTILATION GUIDE RESP GOALS INITIAL MONITORING TARGETS FAILURE SETTINGS 6 P’s BASIC HEMODYNAMIC 1 BLOOD PRESSURE SBP > 90mmHg STABILITY PEAK INSPIRATORY 2 < 35cmH O PRESSURE (PIP) 2 BAROTRAUMA PLATEAU PRESSURE (P ) < 30cmH O PREVENTION PLAT 2 SAFETY SAFETY 3 AutoPEEP None VOLUTRAUMA Start Here TIDAL VOLUME (V ) ~ 6-8cc/kg IBW PREVENTION T Loss of AIRWAY Female ETT 7.0-7.5 AIRWAY / ETT / TRACH Patent Airway MAINTENANCE Male ETT 8.0-8.5 AIRWAY AIRWAY FiO2 21 - 100% PULSE OXIMETRY (SpO2) > 90% Hypoxia OXYGENATION 4 PEEP 5 [5-15] pO2 > 60mmHg 5’5” = 350cc [max 600] pCO2 40mmHg TIDAL 6’0” = 450cc [max 750] 5 VOLUME 6’5” = 500cc [max 850] ETCO2 45 Hypercapnia VENTILATION pH 7.4 GAS GAS EXCHANGE BPM (RR) 14 [10-30] GAS EXCHANGE MINUTE VENTILATION (VMIN) > 5L/min SYNCHRONY WORK OF BREATHING Decreased High Work ASSIST CONTROL MODE VOLUME or PRESSURE of Breathing PATIENT-VENTILATOR AC (V) / AC (P) 6 Comfortable Breaths (WOB) SUPPORT SYNCHRONY COMFORT COMFORT 2⁰ ASSESSMENT PATIENT CIRCUIT VENT Mental Status PIP RR, WOB Pulse, HR, Rhythm ETT/Trach Position Tidal Volume (V ) Trachea T Blood Pressure Secretions Minute Ventilation (V ) SpO MIN Skin Temp/Color 2 Connections Synchrony ETCO Cap Refill 2 Air-Trapping 1. Recognize Signs of Shock Work-up and Manage 2. Assess 6Ps If single problem Troubleshoot Cause 3. If Multiple Problems QUICK FIX Troubleshoot Cause(s) PROBLEMS ©2017 Mayo Clinic Foundation for Medical Education and Research CAUSES QUICK FIX MANAGEMENT Bleeding Hemostasis, Transfuse, Treat cause, Temperature control HYPOVOLEMIA Dehydration Fluid Resuscitation (End points = hypoxia, ↑StO2, ↓PVI) 3rd Spacing Treat cause, Beware of hypoxia (3rd spacing in lungs) Pneumothorax Needle D, Chest tube Abdominal Compartment Syndrome FLUID Treat Cause, Paralyze, Surgery (Open Abdomen) OBSTRUCTED BLOOD RETURN Air-Trapping (AutoPEEP) (if not hypoxic) Pop off vent & SEE SEPARATE CHART PEEP Reduce PEEP Cardiac Tamponade Pericardiocentesis, Drain.