A Proteomic Landscape of Diffuse-Type Gastric Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Propranolol-Mediated Attenuation of MMP-9 Excretion in Infants with Hemangiomas
Supplementary Online Content Thaivalappil S, Bauman N, Saieg A, Movius E, Brown KJ, Preciado D. Propranolol-mediated attenuation of MMP-9 excretion in infants with hemangiomas. JAMA Otolaryngol Head Neck Surg. doi:10.1001/jamaoto.2013.4773 eTable. List of All of the Proteins Identified by Proteomics This supplementary material has been provided by the authors to give readers additional information about their work. © 2013 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 10/01/2021 eTable. List of All of the Proteins Identified by Proteomics Protein Name Prop 12 mo/4 Pred 12 mo/4 Δ Prop to Pred mo mo Myeloperoxidase OS=Homo sapiens GN=MPO 26.00 143.00 ‐117.00 Lactotransferrin OS=Homo sapiens GN=LTF 114.00 205.50 ‐91.50 Matrix metalloproteinase‐9 OS=Homo sapiens GN=MMP9 5.00 36.00 ‐31.00 Neutrophil elastase OS=Homo sapiens GN=ELANE 24.00 48.00 ‐24.00 Bleomycin hydrolase OS=Homo sapiens GN=BLMH 3.00 25.00 ‐22.00 CAP7_HUMAN Azurocidin OS=Homo sapiens GN=AZU1 PE=1 SV=3 4.00 26.00 ‐22.00 S10A8_HUMAN Protein S100‐A8 OS=Homo sapiens GN=S100A8 PE=1 14.67 30.50 ‐15.83 SV=1 IL1F9_HUMAN Interleukin‐1 family member 9 OS=Homo sapiens 1.00 15.00 ‐14.00 GN=IL1F9 PE=1 SV=1 MUC5B_HUMAN Mucin‐5B OS=Homo sapiens GN=MUC5B PE=1 SV=3 2.00 14.00 ‐12.00 MUC4_HUMAN Mucin‐4 OS=Homo sapiens GN=MUC4 PE=1 SV=3 1.00 12.00 ‐11.00 HRG_HUMAN Histidine‐rich glycoprotein OS=Homo sapiens GN=HRG 1.00 12.00 ‐11.00 PE=1 SV=1 TKT_HUMAN Transketolase OS=Homo sapiens GN=TKT PE=1 SV=3 17.00 28.00 ‐11.00 CATG_HUMAN Cathepsin G OS=Homo -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

The Regulation of Interleukin 7 Receptor Alpha Internalization, Recycling and Degradation by IL-7
Universidade de Lisboa Faculdade de Medicina Unidade de Biologia do Cancro, Instituto de Medicina Molecular The regulation of Interleukin 7 receptor alpha internalization, recycling and degradation by IL-7 - Possible implications in T-cell homeostasis, migration and leukaemogenesis - Catarina Martins de Oliveira Henriques Doutoramento em Ciências Biomédicas For the degree of Doctor of Philosophy 2009 Universidade de Lisboa Faculdade de Medicina Unidade de Biologia do Cancro, Instituto de Medicina Molecular The regulation of Interleukin 7 receptor alpha internalization, recycling and degradation by IL-7 - Possible implications in T-cell homeostasis, migration and leukaemogenesis - Catarina Martins de Oliveira Henriques (Recipient of a scholarship- SFRH7BD/21940/2005 from Fundação para a Ciência e Tecnologia) Tese orientada pelo Doutor João T. Barata, Prof Doutor.Gerard Graham e Prof. Doutora Leonor Parreira Doutoramento em Ciências Biomédicas, especialidade em Ciências Biopatológicas For the degree of Doctor of Philosophy 2009 As opiniões expressas são da exclusiva responsabilidade do seu autor A impressão desta dissertação foi aprovada pela Comissão Coordenadora do Conselho Científico da Faculdade de Medicina de Lisboa em reunião de 13 de Outubro de 2009. Para a Prof. Filomena Mota. Sem o seu apoio e inspiração há muitos anos atrás, eu não seria hoje uma bióloga nem esta tese teria alguma vez existido… Table of Contents Table of contents……………………………………………………….………………….................. i Aknowledgements……………………………………………………………………………………………. -

An Attempt to Polarize Human Neutrophils Toward N1 and N2 Phenotypes in Vitro
fimmu-11-00532 April 24, 2020 Time: 17:59 # 1 ORIGINAL RESEARCH published: 28 April 2020 doi: 10.3389/fimmu.2020.00532 An Attempt to Polarize Human Neutrophils Toward N1 and N2 Phenotypes in vitro Mareike Ohms, Sonja Möller and Tamás Laskay* Department of Infectious Diseases and Microbiology, University of Lübeck, Lübeck, Germany Neutrophils act as the first line of defense against invading pathogens. Although traditionally considered in context of their antimicrobial effector functions, the importance of tumor-associated neutrophils (TANs) in the development of cancer has become increasingly clear during the last decade. With regard to their high plasticity, neutrophils were shown to acquire an anti-tumorigenic N1 or a pro-tumorigenic N2 phenotype. Despite the urgent need to get a comprehensive understanding of the interaction of TANs with their tumor microenvironment, most studies still rely on murine tumor models. Here we present for the first time a polarization attempt to generate N1 and N2 neutrophils from primary human neutrophils in vitro. Our results underscore Edited by: that N1-polarized neutrophils have a pro-inflammatory phenotype characterized among Martin Herrmann, University Hospital Erlangen, Germany others by a higher level of intercellular adhesion molecule (ICAM)-1 and high secretion Reviewed by: of interferon (IFN)g-induced protein 10 (IP-10)/C-X-C motif chemokine 10 (CXCL10) Payel Sil, and tumor necrosis factor (TNF). Further, we demonstrate that neutrophils incubated National Institute of Environmental under a tumor-mimicking in vitro environment show a high cell surface expression of Health Sciences (NIEHS), United States C-X-C motif chemokine receptor 2 (CXCR2) and secrete high levels of interleukin (IL)- Mihaela Gadjeva, 8. -

SUPPLEMENTARY METHODS Cell Culture.-Human Peripheral Blood
SUPPLEMENTARY METHODS Cell culture.-Human peripheral blood mononuclear cells (PBMC) were isolated from buffy coats from normal donors over a Lymphoprep (Nycomed Pharma) gradient. Monocytes were purified from PBMC by magnetic cell sorting using CD14 microbeads (Miltenyi Biotech). Monocytes were cultured at 0.5 x 106 cells/ml for 7 days in RPMI 1640 (standard RPMI, which contains 1 mg/L folic acid) supplemented with 10% fetal calf serum, at 37ºC in a humidified atmosphere with 5% CO2, and containing GM-CSF (1000U/ml) or M-CSF (10 ng/ml, ImmunoTools) to generate GM-CSF- polarized macrophages (GM-MØ) or M-CSF-polarized macrophages (M-MØ). MTX pharmacokinetic studies in RA patients administered 25 mg MTX showed peak plasma levels of 1-2 µM MTX two hours after drug administration, but plasma levels decline to 10-50 nM MTX within 24-48 hours [1, 2]. MTX (50 nM), pemetrexed (PMX, 50 nM), folic acid (FA, 50 nM) [3], thymidine (dT, 10 µM), pifithrin-α (PFT, 25-50 µM), nutlin-3 (10 µM, Sigma-Aldrich) was added once on monocytes together with the indicated cytokine, or on monocytes and 7-day differentiated macrophages for 48h. Gene expression profiling.-For long-term MTX treatment, RNA was isolated from three independent preparations of monocytes either unexposed or exposed to MTX (50 nM) and differentiated to GM-MØ or M-MØ for 7-days. For short-term schedule, RNA was isolated from three independent samples of fully differentiated GM-MØ either unexposed or exposed to MTX (50 nM) for 48h, by using RNeasy Mini kit (QIAGEN). -

MALE Protein Name Accession Number Molecular Weight CP1 CP2 H1 H2 PDAC1 PDAC2 CP Mean H Mean PDAC Mean T-Test PDAC Vs. H T-Test
MALE t-test t-test Accession Molecular H PDAC PDAC vs. PDAC vs. Protein Name Number Weight CP1 CP2 H1 H2 PDAC1 PDAC2 CP Mean Mean Mean H CP PDAC/H PDAC/CP - 22 kDa protein IPI00219910 22 kDa 7 5 4 8 1 0 6 6 1 0.1126 0.0456 0.1 0.1 - Cold agglutinin FS-1 L-chain (Fragment) IPI00827773 12 kDa 32 39 34 26 53 57 36 30 55 0.0309 0.0388 1.8 1.5 - HRV Fab 027-VL (Fragment) IPI00827643 12 kDa 4 6 0 0 0 0 5 0 0 - 0.0574 - 0.0 - REV25-2 (Fragment) IPI00816794 15 kDa 8 12 5 7 8 9 10 6 8 0.2225 0.3844 1.3 0.8 A1BG Alpha-1B-glycoprotein precursor IPI00022895 54 kDa 115 109 106 112 111 100 112 109 105 0.6497 0.4138 1.0 0.9 A2M Alpha-2-macroglobulin precursor IPI00478003 163 kDa 62 63 86 72 14 18 63 79 16 0.0120 0.0019 0.2 0.3 ABCB1 Multidrug resistance protein 1 IPI00027481 141 kDa 41 46 23 26 52 64 43 25 58 0.0355 0.1660 2.4 1.3 ABHD14B Isoform 1 of Abhydrolase domain-containing proteinIPI00063827 14B 22 kDa 19 15 19 17 15 9 17 18 12 0.2502 0.3306 0.7 0.7 ABP1 Isoform 1 of Amiloride-sensitive amine oxidase [copper-containing]IPI00020982 precursor85 kDa 1 5 8 8 0 0 3 8 0 0.0001 0.2445 0.0 0.0 ACAN aggrecan isoform 2 precursor IPI00027377 250 kDa 38 30 17 28 34 24 34 22 29 0.4877 0.5109 1.3 0.8 ACE Isoform Somatic-1 of Angiotensin-converting enzyme, somaticIPI00437751 isoform precursor150 kDa 48 34 67 56 28 38 41 61 33 0.0600 0.4301 0.5 0.8 ACE2 Isoform 1 of Angiotensin-converting enzyme 2 precursorIPI00465187 92 kDa 11 16 20 30 4 5 13 25 5 0.0557 0.0847 0.2 0.4 ACO1 Cytoplasmic aconitate hydratase IPI00008485 98 kDa 2 2 0 0 0 0 2 0 0 - 0.0081 - 0.0 -

Neural Immunoglobulin Superfamily Interaction Networks
Available online at www.sciencedirect.com ScienceDirect Neural immunoglobulin superfamily interaction networks 1 ¨ 2 Kai Zinn and Engin Ozkan The immunoglobulin superfamily (IgSF) encompasses mediates protein–protein interactions, and many proteins hundreds of cell surface proteins containing multiple have both Ig domains and FnIII repeats. immunoglobulin-like (Ig) domains. Among these are neural IgCAMs, which are cell adhesion molecules that mediate Many cell surface IgSF proteins are homophilic or het- interactions between cells in the nervous system. IgCAMs in erophilic adhesion molecules (IgCAMs) containing mul- some vertebrate IgSF subfamilies bind to each other tiple Ig domains. IgCAMs mediate interactions among homophilically and heterophilically, forming small interaction neurons and between neurons and glia. Homophilic networks. In Drosophila, a global ‘interactome’ screen IgCAMs include NCAM and L1 in mammals and Fasci- identified two larger networks in which proteins in one IgSF clin II and Dscam in Drosophila. IgCAM-like proteins are subfamily selectively interact with proteins in a different receptors for axon guidance cues such as Netrins and Slits. subfamily. One of these networks, the ‘Dpr-ome’, includes Finally, neuronal transmembrane signal transduction 30 IgSF proteins, each of which is expressed in a unique subset molecules, such as receptor tyrosine kinases and phos- of neurons. Recent evidence shows that one interacting protein phatases, can have IgCAM-like extracellular (XC) pair within the Dpr-ome network is required for development of domains. the brain and neuromuscular system. This mini review does not cover homophilic IgCAMs, Addresses axon guidance receptors, or signaling receptors, but 1 Division of Biology and Biological Engineering, California Institute of focuses on subfamilies of the IgSF that participate in Technology, Pasadena, CA 91125, United States 2 complex networks of interactions. -

CEACAM1 As a Multi-Purpose Target for Cancer Immunotherapy
CEACAM1 as a multi-purpose target for cancer immunotherapy The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Dankner, Matthew, Scott D. Gray-Owen, Yu-Hwa Huang, Richard S. Blumberg, and Nicole Beauchemin. 2017. “CEACAM1 as a multi- purpose target for cancer immunotherapy.” Oncoimmunology 6 (7): e1328336. doi:10.1080/2162402X.2017.1328336. http:// dx.doi.org/10.1080/2162402X.2017.1328336. Published Version doi:10.1080/2162402X.2017.1328336 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:34375168 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA ONCOIMMUNOLOGY 2017, VOL. 6, NO. 7, e1328336 (16 pages) https://doi.org/10.1080/2162402X.2017.1328336 REVIEW CEACAM1 as a multi-purpose target for cancer immunotherapy Matthew Dankner a, Scott D. Gray-Owen b, Yu-Hwa Huangc, Richard S. Blumberg c, and Nicole Beauchemin a aGoodman Cancer Research Centre, McGill University, Montreal, QC, Canada; bDepartment of Molecular Genetics, University of Toronto, Toronto, ON, Canada; cDivision of Gastroenterology, Hepatology, and Endoscopy, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA ABSTRACT ARTICLE HISTORY CEACAM1 is an extensively studied cell surface molecule with established functions in multiple cancer Received 17 February 2017 types, as well as in various compartments of the immune system. Due to its multi-faceted role as a Revised 3 May 2017 recently appreciated immune checkpoint inhibitor and tumor marker, CEACAM1 is an attractive target for Accepted 5 May 2017 cancer immunotherapy. -
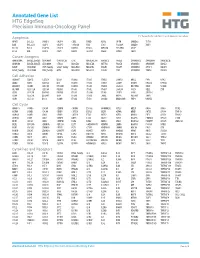
Annotated Gene List HTG Edgeseq Precision Immuno-Oncology Panel
Annotated Gene List HTG EdgeSeq Precision Immuno-Oncology Panel For Research Use Only. Not for use in diagnostic procedures. Apoptosis APAF1 BCL2L1 CARD11 CASP4 CD5L FADD KSR2 OPTN SAMD12 TCF19 BAX BCL2L11 CASP1 CASP5 CORO1A FAS LRG1 PLA2G6 SAMD9 XAF1 BCL10 BCL6 CASP10 CASP8 DAPK2 FASLG MECOM PYCARD SPOP BCL2 BID CASP3 CAV1 DAPL1 GLIPR1 MELK RIPK2 TBK1 Cancer Antigens ANKRD30A BAGE2_BAGE3 CEACAM6 CTAG1A_1B LIPE MAGEA3_A6 MAGEC2 PAGE3 SPANXACD SPANXN4 XAGE1B_1E ARMCX6 BAGE4_BAGE5 CEACAM8 CTAG2 MAGEA1 MAGEA4 MTFR2 PAGE4 SPANXB1 SPANXN5 XAGE2 BAGE CEACAM1 CT45_family GAGE_family MAGEA10 MAGEB2 PAGE1 PAGE5 SPANXN1 SYCP1 XAGE3 BAGE_family CEACAM5 CT47_family HPN MAGEA12 MAGEC1 PAGE2 PBK SPANXN3 TEX14 XAGE5 Cell Adhesion ADAM17 CDH15 CLEC5A DSG3 ICAM2 ITGA5 ITGB2 LAMC3 MBL2 PVR UPK2 ADD2 CDH5 CLEC6A DST ICAM3 ITGA6 ITGB3 LAMP1 MTDH RRAS2 UPK3A ADGRE5 CLDN3 CLEC7A EPCAM ICAM4 ITGAE ITGB4 LGALS1 NECTIN2 SELE VCAM1 ALCAM CLEC12A CLEC9A FBLN1 ITGA1 ITGAL ITGB7 LGALS3 OCLN SELL ZYX CD63 CLEC2B DIAPH3 FXYD5 ITGA2 ITGAM ITLN2 LYVE1 OLR1 SELPLG CD99 CLEC4A DLGAP5 IBSP ITGA3 ITGAX JAML M6PR PECAM1 THY1 CDH1 CLEC4C DSC3 ICAM1 ITGA4 ITGB1 L1CAM MADCAM1 PKP1 UNC5D Cell Cycle ANAPC1 CCND3 CDCA5 CENPH CNNM1 ESCO2 HORMAD2 KIF2C MELK ORC6 SKA3 TPX2 ASPM CCNE1 CDCA8 CENPI CNTLN ESPL1 IKZF1 KIF4A MND1 PATZ1 SP100 TRIP13 AURKA CCNE2 CDK1 CENPL CNTLN ETS1 IKZF2 KIF5C MYBL2 PIF1 SP110 TROAP AURKB CCNF CDK4 CENPU DBF4 ETS2 IKZF3 KIFC1 NCAPG PIMREG SPC24 TUBB BEX1 CDC20 CDK6 CENPW E2F2 EZH2 IKZF4 KNL1 NCAPG2 PKMYT1 SPC25 ZWILCH BEX2 CDC25A CDKN1A CEP250 E2F7 GADD45GIP1 -

A Neutrophil Activation Signature in Covid-19 Athanasios Didangelos
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 20 April 2020 doi:10.20944/preprints202004.0363.v1 A Neutrophil activation signature in Covid-19 Athanasios Didangelos (PhD) [email protected] University of Leicester, Mayer IgA Nephropathy Laboratory, University of Leicester, Leicester, LE1 7RH, United Kingdom Abstract Covid-19 is often related to hyperinflammation that drives lung or multi-organ damage and mortality. The immunopathological mechanisms that cause excessive inflammation following SARS-Cov-2 infection are under investigation while different approaches to limit hyperinflammation in affected patients are being proposed. Here, a computational network approach was used on recently available data to identify possible Covid-19 inflammatory mechanisms. First, network analysis of putative SARS-Cov-2 cellular receptors and their directly associated interacting proteins, led to the mining of a robust neutrophil-response signature and multiple relevant inflammatory response genes. Second, analysis of RNA-seq datasets of lung epithelial cells infected with SARS-Cov-2 found that infected cells specifically expressed neutrophil-attracting chemokines, further supporting the likely role of neutrophils in Covid-19 inflammation. The role of neutrophils in Covid-19 needs to be studied further. Different immunoregulatory molecules and pathways presented here (TNF Receptor, IL8, CXCR1, CXCR2, ADAM10, GPR84, MME-neprilysin, ANPEP, LAP3) are druggable and might be therapeutic targets in efforts to limit SARS-Cov-2 inflammation in severe clinical cases. Introduction New studies have highlighted that Covid-19 is often characterised by an extreme inflammatory response associated with lung and multi-organ injury and mortality and have suggested promising anti-inflammatory options (1). Other studies recommend caution with immunosuppression given that regulated inflammation is necessary for an effective anti-viral response (2). -

Characterizing CEACAM5 Interaction with Cd8α and Cd1d in Intestinal
nature publishing group ARTICLES Characterizing CEACAM5 interaction with CD8a and CD1d in intestinal homeostasis G Roda1, X Jianyu1, MS Park1, L DeMarte2, Z Hovhannisyan1, R Couri1, CP Stanners2, G Yeretssian1 and L Mayer1 Normal intestinal epithelial cells (IECs) could act as non-professional antigen-presenting cells, selectively activating CD8 þ -suppressor T cells. An epithelial cell surface glycoprotein, gp180, recognized by monoclonal antibodies B9 and L12 was determined to be critical in this process. Purification and sequence analysis of mAb B9 reactive material revealed amino-acid sequence homology with CEACAM5. We demonstrate that CEACAM5 has properties attributed to gp180, such as CD8a binding and activation of CD8-associated Lck. CEACAM5 is the only CEACAM member interacting with CD1d through the B3 domain. Its N domain (recognized by B9) is required for CD8a binding. Removal of the N-domain glycosylated residues reduces B9 recognition, CD8a binding affinity, and activation of LcK. Therefore, conformational changes in CEACAM5 glycosylation site are critical for its interaction with CD8a. CEACAM5-activated CD8 þ T cells acquire the ability to suppress the proliferation of CD4 þ T cells in vitro in the presence of interleukin (IL)-15 or IL-7. We provide new insights into the role of CEACAM5 and define its specific immunoregulatory properties among the CEACAMs expressed on IECs. We suggest that unique set of interactions between CEACAM5, CD1d, and CD8 render CD1d more class I-like molecule, facilitating antigen presentation and activation of CD8 þ -suppressor regulatory T cells. INTRODUCTION kinase activity. Subsequently, CD1d becomes more class I-like The nature of the immune response in the intestine may require molecule, with enhanced CD1d binding to the T-cell receptor either immunosuppression or controlled inflammation. -

Mobilisation with G-CSF in Healthy Donors Promotes a High but Temporal Deregulation of Genes
Correspondence 1088 Mobilisation with G-CSF in healthy donors promotes a high but temporal deregulation of genes Leukemia (2005) 19, 1088–1091. doi:10.1038/sj.leu.2403753 version 3.0) and SAM algorithm (‘Significant Analysis of Published online 14 April 2005 Microarrays’). A two-dimensional hierarchical clustering analysis performed TO THE EDITOR on 9203 genes passing the variation filter showed a dendogram with two major branches: one branch included all the donors Over recent years, the use of peripheral blood (PB) instead of after mobilisation with G-CSF, while in the other branch, the bone marrow (BM) as a source for stem cells has been gene profiles from the donors before mobilisation and 2 and 6 increasingly employed both in the autologous and in the months after G-CSF administration were grouped (Figure 1). allogeneic transplant setting. The advantages of peripheral blood When multidimensional scaling analysis was performed, sam- stem cells (PBSC) include easier harvesting of stem cells, ples on day 0 and those on day þ 5 were also assigned into two avoidance of anaesthesia and hospitalisation as well as improved well-differentiated groups. yield of progenitor cells, and a more rapid and robust Genes distinguishing the day 0 samples from the day þ 5 engraftment. However, it should also be taken into account samples were identified as significant by the intersection of data that PBSC mobilisation requires the administration of haemato- sets from SAM analysis. A total of 761 genes distinguished the poietic cytokines (ie granulocyte colony-stimulating factor (G- two groups: 374 increased their expression level, while 387 CSF)) over several days, which stimulate proliferation and decreased their expression level after G-CSF (Table 1).