Highly Efficient Concerted Evolution in the Ribosomal DNA Repeats: Total Rdna Repeat Variation Revealed by Whole-Genome Shotgun Sequence Data
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Database Integrating Genomic Indel Polymorphisms That Distinguish Mouse Strains Keiko Akagi1,2, Robert M
D600–D606 Nucleic Acids Research, 2010, Vol. 38, Database issue Published online 20 November 2009 doi:10.1093/nar/gkp1046 MouseIndelDB: a database integrating genomic indel polymorphisms that distinguish mouse strains Keiko Akagi1,2, Robert M. Stephens3, Jingfeng Li2,4, Evgenji Evdokimov5, Michael R. Kuehn5, Natalia Volfovsky3 and David E. Symer2,4,6,7,8,9,* 1Mouse Cancer Genetics Program, Center for Cancer Research, National Cancer Institute, Frederick, MD 21702, 2Department of Molecular Virology, Immunology and Medical Genetics, The Ohio State University Comprehensive Cancer Center, Columbus, OH 43210, 3Advanced Biomedical Computing Center, Information Systems Program, SAIC-Frederick, Inc., NCI-Frederick, Frederick, MD 21702, 4Basic Research Laboratory, 5Laboratory of Protein Dynamics and Signaling, 6Laboratory of Biochemistry and Molecular Biology, Center for Cancer Research, National Cancer Institute, Frederick, MD 21702, 7Human Cancer Genetics Program, 8Departments of Internal Medicine and 9Biomedical Informatics, The Ohio State University Comprehensive Cancer Center, Columbus, OH 43210, USA Received August 5, 2009; Revised October 23, 2009; Accepted October 27, 2009 ABSTRACT wide-ranging functional differences. This extensive phenotypic variation has helped to make the mouse a MouseIndelDB is an integrated database resource premier model organism, mimicking many aspects of containing thousands of previously unreported human diversity and diseases. Understanding the mouse genomic indel (insertion and deletion) genomic differences that -

Pathway in Protective Immunity Jeremy Manry, Guillaume Laval, Etienne Patin, Simona Fornarino, Christiane Bouchier, Magali Tichit, Luis Barreiro, Lluis Quintana-Murci
Evolutionary genetics evidence of an essential, non-redundant role of the IFN-γ pathway in protective immunity Jeremy Manry, Guillaume Laval, Etienne Patin, Simona Fornarino, Christiane Bouchier, Magali Tichit, Luis Barreiro, Lluis Quintana-Murci To cite this version: Jeremy Manry, Guillaume Laval, Etienne Patin, Simona Fornarino, Christiane Bouchier, et al.. Evo- lutionary genetics evidence of an essential, non-redundant role of the IFN-γ pathway in protective immunity. Human Mutation, Wiley, 2011, 32 (6), pp.633-42. 10.1002/humu.21484. hal-00627541 HAL Id: hal-00627541 https://hal.archives-ouvertes.fr/hal-00627541 Submitted on 29 Sep 2011 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Human Mutation Evolutionary genetics evidence of an essential, non- redundant role of the IFN-γ pathway in protective immunity For Peer Review Journal: Human Mutation Manuscript ID: humu-2010-0462.R1 Wiley - Manuscript type: Research Article Date Submitted by the 23-Jan-2011 Author: Complete List of Authors: Manry, Jeremy; Institut Pasteur Laval, Guillaume; Institut Pasteur Patin, Etienne; Institut Pasteur Fornarino, Simona; Institut Pasteur Bouchier, Christiane; Institut Pasteur Tichit, Magali; Institut Pasteur Barreiro, Luis; University of Chicago Quintana-Murci, Lluis; Institut Pasteur, EEMI IFNG, IFNGR1, IFNGR2, natural selection, polymorphism, Key Words: population genetics John Wiley & Sons, Inc. -
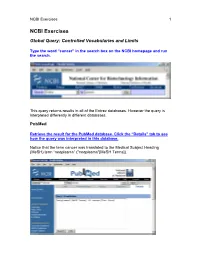
NCBI Exercises 1
NCBI Exercises 1 NCBI Exercises Global Query: Controlled Vocabularies and Limits Type the word “cancer” in the search box on the NCBI homepage and run the search. This query returns results in all of the Entrez databases. However the query is interpreted differently in different databases. PubMed Retrieve the result for the PubMed database. Click the “Details” tab to see how the query was interpreted in this database. Notice that the term cancer was translated to the Medical Subject Heading (MeSH) term “neoplasms” ("neoplasms"[MeSH Terms]). NCBI Exercises 2 MeSH is a controlled vocabulary that is used to index all articles in PubMed. In the details box, edit the query to remove the portion that searched for cancer as a text word and run the search. Notice that the number of articles retrieved has changed. These will be a more relevant set of results. You can force the PubMed engine to only search the MeSH vocabulary or specify any other indexed field through the “Limits” tab. Use the Web browser’s back button to return to the Global query page and retrieve the PubMed results again. Now click on the “Limits” tab. Select “MeSH terms” from the first drop-down menu, the one headed by “All Fields”. Now run the search with the limit in place and check the “Details” tab to verify that only the MeSH term translation was used. Nucleotide Use the Web browser’s back button to return to the Global query page. Retrieve the results for the nucleotide database. Click the “Details” tab to see how the query was interpreted for this molecular database. -

559.Full.Pdf
Copyright 0 1992 by the Genetics Society of America The Structure and Evolutionof Subtelomeric Y‘ Repeats in Saccharomyces cerevisiae Edward J.Louis’ and James E. Haber Rosenstiel Center and Department of Biology, Brandeis University, Waltham, Massachusetts 02254-91 10 Manuscript received September 25, 199 1 Accepted for publication March 28, 1992 ABSTRACT The subtelomeric Y’ family of repeated DNA sequences in the yeast Saccharomyces cerevisiae is of unknown origin and function. Y’s vary in copy number and location among strains. Eight Y‘s, from two strains, were cloned and sequenced over the same 3.2-kb interval in order to assess the within- and between-strain variation as well as address their origin and function. One entireY’ sequence was reconstructed from two clones presented here and apreviously sequenced 833-bp region. It contains two large overlapping open reading frames (ORFs). The putative protein sequences have no strong homologies to any known proteins except for one region that has 27% identity with RNA helicases. RNA homologous to each ORF was detected. Comparison of the sequences revealed that the known long (Y’-L) and short (Y’-S) size classes, which coexist within cells, differ by several insertions and/or deletions within this region. The Y’-Ls from strain Y55 alsodiffer from those of strain YPl by several short deletions in the same region. Most of these deletions appear to have occurred between short (2-10 bp) direct repeats. The single base pair polymorphisms and the deletions are clustered in the first half of the interval compared. There is 0.30-1.13% divergence among Y’-Ls within a strain and 1.15-1.75% divergence between strains in the interval. -

ITS Non-Concerted Evolution and Rampant Hybridization in the Legume Genus Lespedeza
www.nature.com/scientificreports OPEN ITS non-concerted evolution and rampant hybridization in the legume genus Lespedeza Received: 15 August 2016 Accepted: 30 November 2016 (Fabaceae) Published: 04 January 2017 Bo Xu1, Xiao-Mao Zeng1, Xin-Fen Gao1, Dong-Pil Jin2 & Li-Bing Zhang3 The internal transcribed spacer (ITS) as one part of nuclear ribosomal DNA is one of the most extensively sequenced molecular markers in plant systematics. The ITS repeats generally exhibit high-level within-individual homogeneity, while relatively small-scale polymorphism of ITS copies within individuals has often been reported in literature. Here, we identified large-scale polymorphism of ITS copies within individuals in the legume genus Lespedeza (Fabaceae). Divergent paralogs of ITS sequences, including putative pseudogenes, recombinants, and multiple functional ITS copies were sometimes detected in the same individual. Thirty-seven ITS pseudogenes could be easily detected according to nucleotide changes in conserved 5.8S motives, the significantly lower GC contents in at least one of three regions, and the lost ability of 5.8S rDNA sequence to fold into a conserved secondary structure. The distribution patterns of the putative functional clones were highly different between the traditionally recognized two subgenera, suggesting different rates of concerted evolution in two subgenera which could be attributable to their different extents/frequencies of hybridization, confirmed by our analysis of the single-copy nuclear gene PGK. These findings have significant implications in using ITS marker for reconstructing phylogeny and studying hybridization. Concerted evolution is a form of multigene family evolution in which all the tendency of the different genes in a gene family or cluster are assumed to evolve as a unit in concert1,2. -

(12) Patent Application Publication (10) Pub. No.: US 2008/0085284 A1 Patell Et Al
US 20080O85284A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2008/0085284 A1 Patell et al. (43) Pub. Date: Apr. 10, 2008 (54) CONSTRUCTION OF A COMPARATIVE (52) U.S. Cl. ............................. 424/184.1; 435/6: 514/44; DATABASE AND IDENTIFICATION OF 536/23.7:536/24.33: 707/102 VIRULENCE FACTORS COMPARISON OF POLYMORPHC REGIONS IN CLINICAL (57) ABSTRACT SOLATES OF INFECTIOUS ORGANISMS The present invention is directed to novel nucleotide (76) Inventors: Villoo Morawala Patell, Bangalore sequences to be used for diagnosis, identification of the (IN); K.R. Rajyashri, Bangalore (IN); strain, typing of the strain and giving orientation to its Marc Rodrigue, Marcy (FR); Guy potential degree of virulence, infectivity and/or latency for Vernet, Marcy (FR) all infectious diseases more particularly tuberculosis. The present invention also includes method for the identification Correspondence Address: and selection of polymorphisms associated with the viru SALWANCHIK LLOYD & SALWANCHK lence and/or infectivity in infectious diseases more particu A PROFESSIONAL ASSOCATION larly in tuberculosis by a comparative genomic analysis of PO BOX 142950 the sequences of different clinical isolates/strains of infec GAINESVILLE, FL 32614-2950 (US) tious organisms. The regions of polymorphisms, can also act (21) Appl. No.: 11/632,108 as potential drug targets and vaccine targets. More particu larly, the invention also relates to identifying virulence (22) Filed: Apr. 9, 2007 factors of M. tuberculosis strains and other infectious organ isms to be included in a diagnostic DNA chip allowing Publication Classification identification of the strain, typing of the strain and finally (51) Int. Cl. -

The Organization and Evolution of the Responder Satellite in Species Of
Larracuente BMC Evolutionary Biology 2014, 14:233 http://www.biomedcentral.com/1471-2148/14/233 RESEARCH ARTICLE Open Access The organization and evolution of the Responder satellite in species of the Drosophila melanogaster group: dynamic evolution of a target of meiotic drive Amanda M Larracuente Abstract Background: Satellite DNA can make up a substantial fraction of eukaryotic genomes and has roles in genome structure and chromosome segregation. The rapid evolution of satellite DNA can contribute to genomic instability and genetic incompatibilities between species. Despite its ubiquity and its contribution to genome evolution, we currently know little about the dynamics of satellite DNA evolution. The Responder (Rsp) satellite DNA family is found in the pericentric heterochromatin of chromosome 2 of Drosophila melanogaster. Rsp is well-known for being the target of Segregation Distorter (SD)? an autosomal meiotic drive system in D. melanogaster. I present an evolutionary genetic analysis of the Rsp family of repeats in D. melanogaster and its closely-related species in the melanogaster group (D. simulans, D. sechellia, D. mauritiana, D. erecta, and D. yakuba) using a combination of available BAC sequences, whole genome shotgun Sanger reads, Illumina short read deep sequencing, and fluorescence in situ hybridization. Results: I show that Rsp repeats have euchromatic locations throughout the D. melanogaster genome, that Rsp arrays show evidence for concerted evolution, and that Rsp repeats exist outside of D. melanogaster, in the melanogaster group. The repeats in these species are considerably diverged at the sequence level compared to D. melanogaster, and have a strikingly different genomic distribution, even between closely-related sister taxa. -

A Universal Method for Automated Gene Mapping Comment Peder Zipperlen¤*, Knud Nairz¤†, Ivo Rimann†, Konrad Basler*, Ernst Hafen†, Michael Hengartner* and Alex Hajnal†
Open Access Method2005ZipperlenetVolume al. 6, Issue 2, Article R19 A universal method for automated gene mapping comment Peder Zipperlen¤*, Knud Nairz¤†, Ivo Rimann†, Konrad Basler*, Ernst Hafen†, Michael Hengartner* and Alex Hajnal† Addresses: *Institute of Molecular Biology, University of Zürich, Winterthurerstrasse 190, CH-8057 Zürich, Switzerland. †Institute of Zoology, University of Zürich, Winterthurerstrasse 190, CH-8057 Zürich, Switzerland. ¤ These authors contributed equally to this work. reviews Correspondence: Peder Zipperlen. E-mail: [email protected]. Knud Nairz. E-mail: [email protected] Published: 17 January 2005 Received: 9 September 2004 Revised: 15 November 2004 Genome Biology 2005, 6:R19 Accepted: 9 December 2004 The electronic version of this article is the complete one and can be found online at http://genomebiology.com/2005/6/2/R19 reports © 2005 Zipperlen et al.; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Mapping<p>Amaps forhigh-throughput DrosophilaInDel sequence and method C.polymorphisms. elegans.</p> for genotyping by mapping InDels. This method has been used to create fragment-length polymorphism deposited research Abstract Small insertions or deletions (InDels) constitute a ubiquituous class of sequence polymorphisms found in eukaryotic genomes. Here, we present an automated high-throughput genotyping method that relies on the detection of fragment-length polymorphisms (FLPs) caused by InDels. The protocol utilizes standard sequencers and genotyping software. We have established genome-wide FLP maps for both Caenorhabditis elegans and Drosophila melanogaster that facilitate genetic mapping with a minimum of manual input and at comparatively low cost. -

Gene Conversion in Duplicated Genes
Genes 2011, 2, 394-396; doi:10.3390/genes2020394 OPEN ACCESS genes ISSN 2073-4425 www.mdpi.com/journal/genes Editorial Special Issue: Gene Conversion in Duplicated Genes Hideki Innan Graduate University for Advanced Studies, Hayama, Kanagawa 240-0193, Japan; E-Mail: [email protected] Received: 13 June 2011 / Accepted: 17 June 2011 / Published: 17 June 2011 Gene conversion is an outcome of recombination, causing non-reciprocal transfer of a DNA fragment. Several decades later than the discovery of crossing over, gene conversion was first recognized in fungi when non-Mendelian allelic distortion was observed. Gene conversion occurs when a double-strand break is repaired by using homologous sequences in the genome. In meiosis, there is a strong preference to use the orthologous region (allelic gene conversion), which causes non-Mendelian allelic distortion, but paralogous or duplicated regions can also be used for the repair (inter-locus gene conversion, also referred to as non-allelic and ectopic gene conversion). The focus of this special issue is the latter, interlocus gene conversion; the rate is lower than allelic gene conversion but it has more impact on phenotype because more drastic changes in DNA sequence are involved. This special issue consists of 10 review papers covering various aspects of interlocus gene conversion. Hastings [1] provides a review on the basic mechanisms of gene conversion in meiotic cells. Gene conversion occurs through the repair process of a double-strand break with the formation of a D-loop. Gene conversion also occurs in mitosis; the mechanisms should be the same but different proteins may be involved. -

Adriana Antônia Da Cruz Furini Malária Vivax No Estado
Faculdade de Medicina de São José do Rio Preto Programa de Pós-graduação em Ciências da Saúde Adriana Antônia da Cruz Furini Malária vivax no Estado do Pará: influência de polimorfismos nos genes TNFA, IFNG e IL10 associados à resposta imune humoral e ancestralidade genômica. São José do Rio Preto 2016 Adriana Antônia da Cruz Furini Malária vivax no Estado do Pará: influência de polimorfismos nos genes TNFA, IFNG e IL10 associados à resposta imune humoral e ancestralidade genômica. Tese apresentada à Faculdade de Medicina de São José do Rio Preto para obtenção do Título de Doutor no Programa de Pós Graduação em Ciências da Saúde, Eixo Temático: Medicina e Ciências Correlatas. Orientador: Prof. Dr. Ricardo Luiz Dantas Machado São José do Rio Preto 2016 Furini, Adriana Antônia da Cruz Malária vivax no Estado do Pará: influência de polimorfismos nos genes TNFA, IFNG e IL10 associados à resposta imune humoral e ancestralidade genômica./ Adriana Antônia da Cruz Furini São José do Rio Preto, 2016. 124p. Tese (Doutorado) – Faculdade de Medicina de São José do Rio Preto – FAMERP Eixo Temático: Medicina e Ciências Correlatas Orientador: Prof. Dr. Ricardo Luiz Dantas Machado 1.Ancestralidade; 2. Anticorpos; 3. Citocinas; 4. Malária; 5.Plasmodium vivax. ADRIANA ANTÔNIA DA CRUZ FURINI Malária vivax no Estado do Pará: influência de polimorfismos nos genes TNFA, IFNG e IL10 associados à resposta imune humoral e ancestralidade genômica. BANCA EXAMINADORA TESE PARA OBTENÇÃO DO GRAU DE DOUTOR Presidente/Orientador: Prof. Dr. Ricardo Luiz Dantas Machado 2º Examinador:Prof. Dr. Carlos Eugênio Cavasini 3º Examinador: Profa. Dra. Heloísa da Silveira Paro Pedro 4º Examinador: Profa. -

(12) United States Patent (10) Patent No.: US 6,686,163 B2 Allen Et Al
US006686163B2 (12) United States Patent (10) Patent No.: US 6,686,163 B2 Allen et al. (45) Date of Patent: Feb. 3, 2004 (54) CODING SEQUENCE HAPLOTYPE OF THE 5,912,127 A 6/1999 Narod et al. ................... 435/6 HUMAN BRCA1 GENE 5.948,643 A 9/1999 Rubinfeld et al. ......... 435/69.1 5,965,377 A 10/1999 Adams et al. ............. 435/7.23 (75) Inventors: Antonette C. P. Allen, Severn, MD 6,033,857 A 3/2000 Tavtigian et al. .............. 435/6 (US); Tracy S. Angelly, Gaithersburg, 6,045,997 A 4/2000 Futreal et al. - - - - - - - - - - 435/6 MD (US); Tammy Lawrence, Laurel, 6,051,379 A 4/2000 Lescallett et al. .............. 435/6 6,083,698 A 7/2000 Olson et al. ................... 435/6 MD (US); Sheri J.Olson, Falls 6,124,104 A 9/2000 Tavtigian et al. ............ 435/7.2 Church, VA (US); Mark B. Rabin, 6,130,322 A * 10/2000 Murphy et al. ................ 435/6 Rockville, MD (US) FOREIGN PATENT DOCUMENTS (73) Assignee: Gene Logic Inc., Gaithersburg, MD EP O705902 A1 4/1996 (US) EP O705903 A1 4/1996 EP O699.754 A1 6/1996 (*) Notice: Subject to any disclaimer, the term of this GB 2307477 A 5/1997 patent is extended or adjusted under 35 WO WO9304200 3/1993 U.S.C. 154(b) by 0 days. WO WO9519369 7/1995 WO WO9722689 6/1997 WO WO973O108 8/1997 (21) Appl. No.: 10/022,819 WO WO9815654 4/1998 (22) Filed: Dec. 20, 2001 OTHER PUBLICATIONS (65) Prior Publication Data Abeliovich et al. -

Concerted Evolution of Rdna in Recently Formed Tragopogon Allotetraploids Is Typically Associated with an Inverse Correlation Between Gene Copy Number and Expression
Copyright Ó 2007 by the Genetics Society of America DOI: 10.1534/genetics.107.072751 Concerted Evolution of rDNA in Recently Formed Tragopogon Allotetraploids Is Typically Associated With an Inverse Correlation Between Gene Copy Number and Expression Roman Matya´sˇek,* Jennifer A. Tate,† Yoong K. Lim,‡ Hana Sˇrubarˇova´,* Jin Koh,† Andrew R. Leitch,‡ Douglas E. Soltis,† Pamela S. Soltis§ and Alesˇ Kovarˇ´ık*,1 *Institute of Biophysics, Academy of Sciences of the Czech Republic, v.v.i, Laboratory of Molecular Epigenetics, Kra´lovopolska´ 135, CZ-61265 Brno, Czech Republic, †Department of Botany, University of Florida, Gainesville, Florida 32611, ‡School of Biological Sciences, Queen Mary, University of London, E1 4NS, UK and §Florida Museum of Natural History, University of Florida, Gainesville, Florida 32611 Manuscript received March 1, 2007 Accepted for publication May 23, 2007 ABSTRACT We analyzed nuclear ribosomal DNA (rDNA) transcription and chromatin condensation in individuals from several populations of Tragopogon mirus and T. miscellus, allotetraploids that have formed repeatedly within only the last 80 years from T. dubius and T. porrifolius and T. dubius and T. pratensis, respectively. We identified populations with no (2), partial (2), and complete (4) nucleolar dominance. It is probable that epigenetic regulation following allopolyploidization varies between populations, with a tendency toward nucleolar dominance by one parental homeologue. Dominant rDNA loci are largely decondensed at interphase while silent loci formed condensed heterochromatic regions excluded from nucleoli. Those populations where nucleolar dominance is fixed are epigenetically more stable than those with partial or incomplete dominance. Previous studies indicated that concerted evolution has partially homogenized thousands of parental rDNA units typically reducing the copy numbers of those derived from the T.