Transition Bias Influences the Evolution of Antibiotic Resistance in Mycobacterium
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Analysis of the Transition from Wild to Domesticated Cotton (G
bioRxiv preprint doi: https://doi.org/10.1101/616763; this version posted April 23, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Genetic analysis of the transition from wild to domesticated cotton (G. hirsutum) Corrinne E. Grover1, Mi-Jeong Yoo1,a, Michael A. Gore2, David B. Harker3,b, Robert L. Byers3, Alexander E. Lipka4, Guanjing Hu1, Daojun Yuan1, Justin L. Conover1, Joshua A. Udall3,c, Andrew H. Paterson5, and Jonathan F. Wendel1 1Department of Ecology, Evolution and Organismal Biology, Iowa State University, Ames, IA 50011 2Plant Breeding and Genetics Section, School of Integrative Plant Science, Cornell University, Ithaca, NY 14853 3Plant and Wildlife Science Department, Brigham Young University, Provo, UT 84602 4Department of Crop Sciences, University of Illinois, Urbana, IL 61801 5Plant Genome Mapping Laboratory, University of Georgia, Athens, GA 30602 acurrent address: Department of Biology, University of Florida, Gainesville, FL 32611 bcurrent address: Department of Dermatology, The University of Texas Southwestern Medical Center, Dallas, TX 75390 ccurrent address: Southern Plains Agricultural Research Center, USDA-ARS, College Station, TX, 77845- 4988 Corresponding Author: Jonathan F. Wendel ([email protected]) Data Availability: All data are available via figshare (https://figshare.com/projects/Genetic_analysis_of_the_transition_from_wild_to_domesticated_cotton_G _hirsutum_QTL/62693) and GitHub (https://github.com/Wendellab/QTL_TxMx). Keywords: QTL, domestication, Gossypium hirsutum, cotton Summary: An F2 population between truly wild and domesticated cotton was used to identify QTL associated with selection under domestication. -

A Database Integrating Genomic Indel Polymorphisms That Distinguish Mouse Strains Keiko Akagi1,2, Robert M
D600–D606 Nucleic Acids Research, 2010, Vol. 38, Database issue Published online 20 November 2009 doi:10.1093/nar/gkp1046 MouseIndelDB: a database integrating genomic indel polymorphisms that distinguish mouse strains Keiko Akagi1,2, Robert M. Stephens3, Jingfeng Li2,4, Evgenji Evdokimov5, Michael R. Kuehn5, Natalia Volfovsky3 and David E. Symer2,4,6,7,8,9,* 1Mouse Cancer Genetics Program, Center for Cancer Research, National Cancer Institute, Frederick, MD 21702, 2Department of Molecular Virology, Immunology and Medical Genetics, The Ohio State University Comprehensive Cancer Center, Columbus, OH 43210, 3Advanced Biomedical Computing Center, Information Systems Program, SAIC-Frederick, Inc., NCI-Frederick, Frederick, MD 21702, 4Basic Research Laboratory, 5Laboratory of Protein Dynamics and Signaling, 6Laboratory of Biochemistry and Molecular Biology, Center for Cancer Research, National Cancer Institute, Frederick, MD 21702, 7Human Cancer Genetics Program, 8Departments of Internal Medicine and 9Biomedical Informatics, The Ohio State University Comprehensive Cancer Center, Columbus, OH 43210, USA Received August 5, 2009; Revised October 23, 2009; Accepted October 27, 2009 ABSTRACT wide-ranging functional differences. This extensive phenotypic variation has helped to make the mouse a MouseIndelDB is an integrated database resource premier model organism, mimicking many aspects of containing thousands of previously unreported human diversity and diseases. Understanding the mouse genomic indel (insertion and deletion) genomic differences that -

Pathway in Protective Immunity Jeremy Manry, Guillaume Laval, Etienne Patin, Simona Fornarino, Christiane Bouchier, Magali Tichit, Luis Barreiro, Lluis Quintana-Murci
Evolutionary genetics evidence of an essential, non-redundant role of the IFN-γ pathway in protective immunity Jeremy Manry, Guillaume Laval, Etienne Patin, Simona Fornarino, Christiane Bouchier, Magali Tichit, Luis Barreiro, Lluis Quintana-Murci To cite this version: Jeremy Manry, Guillaume Laval, Etienne Patin, Simona Fornarino, Christiane Bouchier, et al.. Evo- lutionary genetics evidence of an essential, non-redundant role of the IFN-γ pathway in protective immunity. Human Mutation, Wiley, 2011, 32 (6), pp.633-42. 10.1002/humu.21484. hal-00627541 HAL Id: hal-00627541 https://hal.archives-ouvertes.fr/hal-00627541 Submitted on 29 Sep 2011 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Human Mutation Evolutionary genetics evidence of an essential, non- redundant role of the IFN-γ pathway in protective immunity For Peer Review Journal: Human Mutation Manuscript ID: humu-2010-0462.R1 Wiley - Manuscript type: Research Article Date Submitted by the 23-Jan-2011 Author: Complete List of Authors: Manry, Jeremy; Institut Pasteur Laval, Guillaume; Institut Pasteur Patin, Etienne; Institut Pasteur Fornarino, Simona; Institut Pasteur Bouchier, Christiane; Institut Pasteur Tichit, Magali; Institut Pasteur Barreiro, Luis; University of Chicago Quintana-Murci, Lluis; Institut Pasteur, EEMI IFNG, IFNGR1, IFNGR2, natural selection, polymorphism, Key Words: population genetics John Wiley & Sons, Inc. -
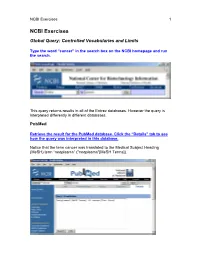
NCBI Exercises 1
NCBI Exercises 1 NCBI Exercises Global Query: Controlled Vocabularies and Limits Type the word “cancer” in the search box on the NCBI homepage and run the search. This query returns results in all of the Entrez databases. However the query is interpreted differently in different databases. PubMed Retrieve the result for the PubMed database. Click the “Details” tab to see how the query was interpreted in this database. Notice that the term cancer was translated to the Medical Subject Heading (MeSH) term “neoplasms” ("neoplasms"[MeSH Terms]). NCBI Exercises 2 MeSH is a controlled vocabulary that is used to index all articles in PubMed. In the details box, edit the query to remove the portion that searched for cancer as a text word and run the search. Notice that the number of articles retrieved has changed. These will be a more relevant set of results. You can force the PubMed engine to only search the MeSH vocabulary or specify any other indexed field through the “Limits” tab. Use the Web browser’s back button to return to the Global query page and retrieve the PubMed results again. Now click on the “Limits” tab. Select “MeSH terms” from the first drop-down menu, the one headed by “All Fields”. Now run the search with the limit in place and check the “Details” tab to verify that only the MeSH term translation was used. Nucleotide Use the Web browser’s back button to return to the Global query page. Retrieve the results for the nucleotide database. Click the “Details” tab to see how the query was interpreted for this molecular database. -

Genetic Testing: Background and Policy Issues
Genetic Testing: Background and Policy Issues Amanda K. Sarata Specialist in Health Policy March 2, 2015 Congressional Research Service 7-5700 www.crs.gov RL33832 Genetic Testing: Background and Policy Issues Summary Congress has considered, at various points in time, numerous pieces of legislation that relate to genetic and genomic technology and testing. These include bills addressing genetic discrimination in health insurance and employment; precision medicine; the patenting of genetic material; and the oversight of clinical laboratory tests (in vitro diagnostics), including genetic tests. The focus on these issues signals the growing importance of public policy issues surrounding the clinical and public health implications of new genetic technology. As genetic technologies proliferate and are increasingly used to guide clinical treatment, these public policy issues are likely to continue to garner attention. Understanding the basic scientific concepts underlying genetics and genetic testing may help facilitate the development of more effective public policy in this area. Humans have 23 pairs of chromosomes in the nucleus of most cells in their bodies. Chromosomes are composed of deoxyribonucleic acid (DNA) and protein. DNA is composed of complex chemical substances called bases. Proteins are fundamental components of all living cells, and include enzymes, structural elements, and hormones. A gene is the section of DNA that contains the sequence which corresponds to a specific protein. Though most of the genome is similar between individuals, there can be significant variation in physical appearance or function between individuals due to variations in DNA sequence that may manifest as changes in the protein, which affect the protein’s function. -

(12) Patent Application Publication (10) Pub. No.: US 2008/0085284 A1 Patell Et Al
US 20080O85284A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2008/0085284 A1 Patell et al. (43) Pub. Date: Apr. 10, 2008 (54) CONSTRUCTION OF A COMPARATIVE (52) U.S. Cl. ............................. 424/184.1; 435/6: 514/44; DATABASE AND IDENTIFICATION OF 536/23.7:536/24.33: 707/102 VIRULENCE FACTORS COMPARISON OF POLYMORPHC REGIONS IN CLINICAL (57) ABSTRACT SOLATES OF INFECTIOUS ORGANISMS The present invention is directed to novel nucleotide (76) Inventors: Villoo Morawala Patell, Bangalore sequences to be used for diagnosis, identification of the (IN); K.R. Rajyashri, Bangalore (IN); strain, typing of the strain and giving orientation to its Marc Rodrigue, Marcy (FR); Guy potential degree of virulence, infectivity and/or latency for Vernet, Marcy (FR) all infectious diseases more particularly tuberculosis. The present invention also includes method for the identification Correspondence Address: and selection of polymorphisms associated with the viru SALWANCHIK LLOYD & SALWANCHK lence and/or infectivity in infectious diseases more particu A PROFESSIONAL ASSOCATION larly in tuberculosis by a comparative genomic analysis of PO BOX 142950 the sequences of different clinical isolates/strains of infec GAINESVILLE, FL 32614-2950 (US) tious organisms. The regions of polymorphisms, can also act (21) Appl. No.: 11/632,108 as potential drug targets and vaccine targets. More particu larly, the invention also relates to identifying virulence (22) Filed: Apr. 9, 2007 factors of M. tuberculosis strains and other infectious organ isms to be included in a diagnostic DNA chip allowing Publication Classification identification of the strain, typing of the strain and finally (51) Int. Cl. -

A Universal Method for Automated Gene Mapping Comment Peder Zipperlen¤*, Knud Nairz¤†, Ivo Rimann†, Konrad Basler*, Ernst Hafen†, Michael Hengartner* and Alex Hajnal†
Open Access Method2005ZipperlenetVolume al. 6, Issue 2, Article R19 A universal method for automated gene mapping comment Peder Zipperlen¤*, Knud Nairz¤†, Ivo Rimann†, Konrad Basler*, Ernst Hafen†, Michael Hengartner* and Alex Hajnal† Addresses: *Institute of Molecular Biology, University of Zürich, Winterthurerstrasse 190, CH-8057 Zürich, Switzerland. †Institute of Zoology, University of Zürich, Winterthurerstrasse 190, CH-8057 Zürich, Switzerland. ¤ These authors contributed equally to this work. reviews Correspondence: Peder Zipperlen. E-mail: [email protected]. Knud Nairz. E-mail: [email protected] Published: 17 January 2005 Received: 9 September 2004 Revised: 15 November 2004 Genome Biology 2005, 6:R19 Accepted: 9 December 2004 The electronic version of this article is the complete one and can be found online at http://genomebiology.com/2005/6/2/R19 reports © 2005 Zipperlen et al.; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Mapping<p>Amaps forhigh-throughput DrosophilaInDel sequence and method C.polymorphisms. elegans.</p> for genotyping by mapping InDels. This method has been used to create fragment-length polymorphism deposited research Abstract Small insertions or deletions (InDels) constitute a ubiquituous class of sequence polymorphisms found in eukaryotic genomes. Here, we present an automated high-throughput genotyping method that relies on the detection of fragment-length polymorphisms (FLPs) caused by InDels. The protocol utilizes standard sequencers and genotyping software. We have established genome-wide FLP maps for both Caenorhabditis elegans and Drosophila melanogaster that facilitate genetic mapping with a minimum of manual input and at comparatively low cost. -

Adriana Antônia Da Cruz Furini Malária Vivax No Estado
Faculdade de Medicina de São José do Rio Preto Programa de Pós-graduação em Ciências da Saúde Adriana Antônia da Cruz Furini Malária vivax no Estado do Pará: influência de polimorfismos nos genes TNFA, IFNG e IL10 associados à resposta imune humoral e ancestralidade genômica. São José do Rio Preto 2016 Adriana Antônia da Cruz Furini Malária vivax no Estado do Pará: influência de polimorfismos nos genes TNFA, IFNG e IL10 associados à resposta imune humoral e ancestralidade genômica. Tese apresentada à Faculdade de Medicina de São José do Rio Preto para obtenção do Título de Doutor no Programa de Pós Graduação em Ciências da Saúde, Eixo Temático: Medicina e Ciências Correlatas. Orientador: Prof. Dr. Ricardo Luiz Dantas Machado São José do Rio Preto 2016 Furini, Adriana Antônia da Cruz Malária vivax no Estado do Pará: influência de polimorfismos nos genes TNFA, IFNG e IL10 associados à resposta imune humoral e ancestralidade genômica./ Adriana Antônia da Cruz Furini São José do Rio Preto, 2016. 124p. Tese (Doutorado) – Faculdade de Medicina de São José do Rio Preto – FAMERP Eixo Temático: Medicina e Ciências Correlatas Orientador: Prof. Dr. Ricardo Luiz Dantas Machado 1.Ancestralidade; 2. Anticorpos; 3. Citocinas; 4. Malária; 5.Plasmodium vivax. ADRIANA ANTÔNIA DA CRUZ FURINI Malária vivax no Estado do Pará: influência de polimorfismos nos genes TNFA, IFNG e IL10 associados à resposta imune humoral e ancestralidade genômica. BANCA EXAMINADORA TESE PARA OBTENÇÃO DO GRAU DE DOUTOR Presidente/Orientador: Prof. Dr. Ricardo Luiz Dantas Machado 2º Examinador:Prof. Dr. Carlos Eugênio Cavasini 3º Examinador: Profa. Dra. Heloísa da Silveira Paro Pedro 4º Examinador: Profa. -

Characterization of Point Mutations in the Same Arginine Codon in Three Unrelated Patients with Ornithine Transcarbamylase Deficiency
Characterization of point mutations in the same arginine codon in three unrelated patients with ornithine transcarbamylase deficiency. A Maddalena, … , W E O'Brien, R L Nussbaum J Clin Invest. 1988;82(4):1353-1358. https://doi.org/10.1172/JCI113738. Research Article Point mutations in the X-linked ornithine transcarbamylase (OTC) gene have been detected at the same Taq I restriction site in 3 of 24 unrelated probands with OTC deficiency. A de novo mutation could be traced in all three families to an individual in a prior generation, confirming independent recurrence. The DNA sequence in the region of the altered Taq I site was determined in the three probands. In two unrelated male probands with neonatal onset of severe OTC deficiency, a guanine (G) to adenine (A) mutation on the sense strand (antisense cytosine [C] to thymine [T]) was found, resulting in glutamine for arginine at amino acid 109 of the mature polypeptide. In the third case, where the proband was a symptomatic female, C to T (sense strand) transition converted residue 109 to a premature stop. These results support the observation that Taq I restriction sites, which contain an internal CG, are particularly susceptible to C to T transition mutation due to deamination of a methylated C in either the sense or antisense strand. The OTC gene seems especially sensitive to C to T transition mutation at arginine codon 109 because either a nonsense mutation or an extremely deleterious missense mutation will result. Find the latest version: https://jci.me/113738/pdf Characterization of Point Mutations in the Same Arginine Codon in Three Unrelated Patients with Ornithine Transcarbamylase Deficiency Anne Maddalena,*" J. -

Indel Information Eliminates Trivial Sequence Alignment in Maximum Likelihood Phylogenetic Analysis
Cladistics Cladistics 28 (2012) 514–528 10.1111/j.1096-0031.2012.00402.x Indel information eliminates trivial sequence alignment in maximum likelihood phylogenetic analysis John S.S. Dentona,b,* and Ward C. Wheelerb,c aDivision of Vertebrate Zoology, American Museum of Natural History, New York, NY 10024, USA; bRichard Gilder Graduate School, American Museum of Natural History, New York, NY 10024, USA; cDivision of Invertebrate Zoology, American Museum of Natural History, New York, NY 10024, USA Accepted 12 March 2012 Abstract Although there has been a recent proliferation in maximum-likelihood (ML)-based tree estimation methods based on a fixed sequence alignment (MSA), little research has been done on incorporating indel information in this traditional framework. We show, using a simple model on a single character example, that a trivial alignment of a different form than that previously identified for parsimony is optimal in ML under standard assumptions treating indels as ‘‘missing’’ data, but that it is not optimal when indels are incorporated into the character alphabet. We show that the optimality of the trivial alignment is not an artefact of simplified theory assumptions by demonstrating that trivial alignment likelihoods of five different multiple sequence alignment datasets exhibit this phenomenon. These results demonstrate the need for use of indel information in likelihood analysis on fixed MSAs, and suggest that caution must be exercised when drawing conclusions from software implementations claiming improvements in likelihood scores under an indels-as-missing assumption. Ó The Willi Hennig Society 2012. Maximum likelihood (ML) has become a popular the increasing sophistication of search heuristics in ML optimality criterion for inferring evolutionary trees software (e.g. -
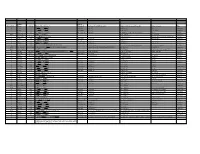
Systematic (Genomic Based)
Trivial Codon Exon/Intron Name Codon Nucleotide Change* Changed Systematic(cDNA based) Systematic (genomic based) Other names Type 2 M-1T -1 CCATGGC>CCACGGC Met>Thr c.2T>C g.4895T>C M1T transition 2 R3Op 3 CACCGAT>CACTGAT Arg>Opal c.10C>T g.4903C>T R3X, R3ter transition 2 ∆A20 20 CCC[A]GAG>CCCGAG Gln>Arg (fs) c.62delA g.4955delA (first base of deletion) ∆A20 deletion IVS2 IVS2-1G>A atagGTA>ataaGTA g.5814G>A c.113-1G>A transition 3 IVS2-1∆4 37-38 ata[gGTA]CCA>ataCCA g.5813delGGTA G12/∆3;IVS2-delGGTA;∆4IVS2-E3 deletion 3 R59op 59 TTCCGAG>TTCTGAG Arg>Opal c.178C>T g.5880C>T R59X;R59ter transition 3 I73T 73 GCATCGG>GCACCGG Ile>Thr c.221T>C g.5923T>C transition 3 L83∆C 83 CCT[C]TAC>CCTTAC Leu>Leu(fs) c.252delC g.5954delC c.250 delC deletion 3 1123ins12 104 GGT[]GGG>GGT[GGGGATCGTGGT]GGG c.314^315ins12 g.6016^6017insGGGGATCGTGGT —12E3;V104+GIVV insertion IVS3 K107K∆12 104-107del GTG[GTGGGAATCAAG]gtt>GTGgtgggaatcaaagtt c.324G>A;delGTGGGAATCAAG(312-324) g.6026G>A g.1133G>A transition IVS3 IVS3-1G>A acagTTA>acaaTTA g.7257G>A c.325-1G>C, g.8180G>C,IVS3sas transversion 4 ∆28E4 114-151 TCC[TCTTGCAGGAACAAACAAAGAAACCACC]ATT>TCCATT Pro>Pro(fs) c.345-372del28 g.7277^7305del c.345_72del28 deletion 4 Q110Oc 110 GACCAAG>GACTAAG Gln>Ochre c.331C>T g.7264C>T transition 4 ∆4E4 118-120 GAAC[AAAC]AAAG>GAACAAAG c.357delAAAC g.7289delAAAC ∆4, MD∆4 deletion 4-5 ∆E4-E5 ccc[tgt…TCC]AGC>cccAGC g.6594^8239del F13/∆4,5 deletion 5 C134R 134 CGCTGTG>CGCCGTG Cys>Arg c.403T>C g.8162T>C C135R transition 5 W147R 147 AAGTGGC>AAGCGGC Trp>Arg c.442T>C g.8201T>C W148R transition -

(12) United States Patent (10) Patent No.: US 6,686,163 B2 Allen Et Al
US006686163B2 (12) United States Patent (10) Patent No.: US 6,686,163 B2 Allen et al. (45) Date of Patent: Feb. 3, 2004 (54) CODING SEQUENCE HAPLOTYPE OF THE 5,912,127 A 6/1999 Narod et al. ................... 435/6 HUMAN BRCA1 GENE 5.948,643 A 9/1999 Rubinfeld et al. ......... 435/69.1 5,965,377 A 10/1999 Adams et al. ............. 435/7.23 (75) Inventors: Antonette C. P. Allen, Severn, MD 6,033,857 A 3/2000 Tavtigian et al. .............. 435/6 (US); Tracy S. Angelly, Gaithersburg, 6,045,997 A 4/2000 Futreal et al. - - - - - - - - - - 435/6 MD (US); Tammy Lawrence, Laurel, 6,051,379 A 4/2000 Lescallett et al. .............. 435/6 6,083,698 A 7/2000 Olson et al. ................... 435/6 MD (US); Sheri J.Olson, Falls 6,124,104 A 9/2000 Tavtigian et al. ............ 435/7.2 Church, VA (US); Mark B. Rabin, 6,130,322 A * 10/2000 Murphy et al. ................ 435/6 Rockville, MD (US) FOREIGN PATENT DOCUMENTS (73) Assignee: Gene Logic Inc., Gaithersburg, MD EP O705902 A1 4/1996 (US) EP O705903 A1 4/1996 EP O699.754 A1 6/1996 (*) Notice: Subject to any disclaimer, the term of this GB 2307477 A 5/1997 patent is extended or adjusted under 35 WO WO9304200 3/1993 U.S.C. 154(b) by 0 days. WO WO9519369 7/1995 WO WO9722689 6/1997 WO WO973O108 8/1997 (21) Appl. No.: 10/022,819 WO WO9815654 4/1998 (22) Filed: Dec. 20, 2001 OTHER PUBLICATIONS (65) Prior Publication Data Abeliovich et al.