Potential Therapeutic Approaches for Modulating Expression and Accumulation of Defective Lamin a in Laminopathies and Age-Related Diseases
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
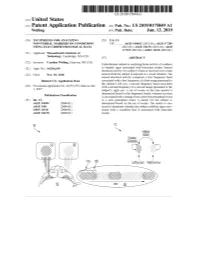
ANNNNNNNNNNNNNNNNNNNN 100A 006 Left Eye Input Right Eye Input
US 20190175049A1 ( 19) United States (12 ) Patent Application Publication (10 ) Pub. No. : US 2019 /0175049 A1 Welling ( 43 ) Pub . Date : Jun . 13 , 2019 ( 54 ) TECHNIQUES FOR ANALYZING (52 ) U . S . CI. NON -VERBAL MARKERS OF CONDITIONS CPC . .. A61B 5 /04842 (2013 . 01 ) ; A61B 5 / 7289 USING ELECTROPHYSIOLOGICAL DATA (2013 . 01) ; A61B 5 /0478 ( 2013 .01 ) ; A61B 5 /7225 ( 2013. 01 ) ; G06N 20 / 10 (2019 .01 ) (71 ) Applicant: Massachusetts Institute of Technology , Cambridge , MA (US ) ( 57 ) ABSTRACT (72 ) Inventor : Caroline Welling, Hanover, NH (US ) Embodiments related to analyzing brain activity of a subject to identify signs associated with binocular rivalry . Sensed ( 21 ) Appl. No. : 16 / 206, 639 electrical activity of a subject' s brain is received over a time period while the subject is exposed to a visual stimulus. The ( 22 ) Filed : Nov. 30 , 2018 sensed electrical activity comprises a first frequency band Related U . S . Application Data associated with a first frequency of a first image presented to the subject ' s left eye , a second frequency band associated (60 ) Provisional application No .62 / 593 , 535, filed on Dec . with a second frequency of a second image presented to the 1 , 2017 subject ' s right eye . A set of events in the time period is determined based on the frequency bands, wherein an event Publication Classification is associated with a change from a previous perceptual event (51 ) Int. Ci. to a new perceptual event. A metric for the subject is A61B 5 /0484 ( 2006 .01 ) determined based on the set of events . The metric is ana A61B 5 /00 ( 2006 .01 ) lyzed to determine whether the subject exhibits signs asso GO6N 20 / 10 (2006 .01 ) ciated with a condition that is associated with binocular A61B 5 /0478 ( 2006 .01 ) rivalry . -

Campro Catalog Stable Isotope
Introduction & Welcome Dear Valued Customer, We are pleased to present to you our Stable Isotopes Catalog which contains more than three thousand (3000) high quality labeled compounds. You will find new additions that are beneficial for your research. Campro Scientific is proud to work together with Isotec, Inc. for the distribution and marketing of their stable isotopes. We have been working with Isotec for more than twenty years and know that their products meet the highest standard. Campro Scientific was founded in 1981 and we provide services to some of the most prestigious universities, research institutes and laboratories throughout Europe. We are a research-oriented company specialized in supporting the requirements of the scientific community. We are the exclusive distributor of some of the world’s leading producers of research chemicals, radioisotopes, stable isotopes and environmental standards. We understand the requirements of our customers, and work every day to fulfill them. In working with us you are guaranteed to receive: - Excellent customer service - High quality products - Dependable service - Efficient distribution The highly educated staff at Campro’s headquarters and sales office is ready to assist you with your questions and product requirements. Feel free to call us at any time. Sincerely, Dr. Ahmad Rajabi General Manager 180/280 = unlabeled 185/285 = 15N labeled 181/281 = double labeled (13C+15N, 13C+D, 15N+18O etc.) 186/286 = 12C labeled 182/282 = d labeled 187/287 = 17O labeled 183/283 = 13C labeleld 188/288 = 18O labeled 184/284 = 16O labeled, 14N labeled 189/289 = Noble Gases Table of Contents Ordering Information.................................................................................................. page 4 - 5 Packaging Information .............................................................................................. -

Regulatory Effects of Caffeic Acid Phenethyl Ester on Neuroinflammation in Microglial Cells
Int. J. Mol. Sci. 2015, 16, 5572-5589; doi:10.3390/ijms16035572 OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Article Regulatory Effects of Caffeic Acid Phenethyl Ester on Neuroinflammation in Microglial Cells Cheng-Fang Tsai 1,†, Yueh-Hsiung Kuo 1,2,†, Wei-Lan Yeh 3, Caren Yu-Ju Wu 4, Hsiao-Yun Lin 5, 4 4 4 1 5,6, Sheng-Wei Lai , Yu-Shu Liu , Ling-Hsuan Wu , Jheng-Kun Lu and Dah-Yuu Lu * 1 Department of Biotechnology, Asia University, Taichung 413, Taiwan; E-Mails: [email protected] (C.-F.T.); [email protected] (Y.-H.K.); [email protected] (J.-K.L.) 2 Department of Chinese Pharmaceutical Sciences and Chinese Medicine Resources, China Medical University, Taichung 404, Taiwan 3 Department of Cell and Tissue Engineering, Changhua Christian Hospital, Changhua 500, Taiwan; E-Mail: [email protected] 4 Graduate Institute of Basic Medical Science, College of Medicine, China Medical University, Taichung 404, Taiwan; E-Mails: [email protected] (C.Y.-J.W.); [email protected] (S.-W.L.); [email protected] (Y.-S.L.); [email protected] (L.-H.W.) 5 Graduate Institute of Neural and Cognitive Sciences, China Medical University, Taichung 404, Taiwan; E-Mail: [email protected] 6 Department of Photonics and Communication Engineering, Asia University, Taichung 413, Taiwan † These authors contributed equally to this work. * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +886-4-2205-3366 (ext. 8206); Fax: +886-4-2207-1507. Academic Editor: Guido R. -

Drug Name Plate Number Well Location % Inhibition, Screen Axitinib 1 1 20 Gefitinib (ZD1839) 1 2 70 Sorafenib Tosylate 1 3 21 Cr
Drug Name Plate Number Well Location % Inhibition, Screen Axitinib 1 1 20 Gefitinib (ZD1839) 1 2 70 Sorafenib Tosylate 1 3 21 Crizotinib (PF-02341066) 1 4 55 Docetaxel 1 5 98 Anastrozole 1 6 25 Cladribine 1 7 23 Methotrexate 1 8 -187 Letrozole 1 9 65 Entecavir Hydrate 1 10 48 Roxadustat (FG-4592) 1 11 19 Imatinib Mesylate (STI571) 1 12 0 Sunitinib Malate 1 13 34 Vismodegib (GDC-0449) 1 14 64 Paclitaxel 1 15 89 Aprepitant 1 16 94 Decitabine 1 17 -79 Bendamustine HCl 1 18 19 Temozolomide 1 19 -111 Nepafenac 1 20 24 Nintedanib (BIBF 1120) 1 21 -43 Lapatinib (GW-572016) Ditosylate 1 22 88 Temsirolimus (CCI-779, NSC 683864) 1 23 96 Belinostat (PXD101) 1 24 46 Capecitabine 1 25 19 Bicalutamide 1 26 83 Dutasteride 1 27 68 Epirubicin HCl 1 28 -59 Tamoxifen 1 29 30 Rufinamide 1 30 96 Afatinib (BIBW2992) 1 31 -54 Lenalidomide (CC-5013) 1 32 19 Vorinostat (SAHA, MK0683) 1 33 38 Rucaparib (AG-014699,PF-01367338) phosphate1 34 14 Lenvatinib (E7080) 1 35 80 Fulvestrant 1 36 76 Melatonin 1 37 15 Etoposide 1 38 -69 Vincristine sulfate 1 39 61 Posaconazole 1 40 97 Bortezomib (PS-341) 1 41 71 Panobinostat (LBH589) 1 42 41 Entinostat (MS-275) 1 43 26 Cabozantinib (XL184, BMS-907351) 1 44 79 Valproic acid sodium salt (Sodium valproate) 1 45 7 Raltitrexed 1 46 39 Bisoprolol fumarate 1 47 -23 Raloxifene HCl 1 48 97 Agomelatine 1 49 35 Prasugrel 1 50 -24 Bosutinib (SKI-606) 1 51 85 Nilotinib (AMN-107) 1 52 99 Enzastaurin (LY317615) 1 53 -12 Everolimus (RAD001) 1 54 94 Regorafenib (BAY 73-4506) 1 55 24 Thalidomide 1 56 40 Tivozanib (AV-951) 1 57 86 Fludarabine -

5994392 Tion of Application No. 67375.734 Eb3-1685, PEN. T
USOO5994392A United States Patent (19) 11 Patent Number: 5,994,392 Shashoua (45) Date of Patent: Nov.30, 1999 54 ANTIPSYCHOTIC PRODRUGS COMPRISING 5,120,760 6/1992 Horrobin ................................. 514/458 AN ANTIPSYCHOTICAGENT COUPLED TO 5,141,958 8/1992 Crozier-Willi et al. ................ 514/558 AN UNSATURATED FATTY ACID 5,216,023 6/1993 Literati et al. .......................... 514/538 5,246,726 9/1993 Horrobin et al. ....................... 424/646 5,516,800 5/1996 Horrobin et al. ....................... 514/560 75 Inventor: Victor E. Shashoua, Brookline, Mass. 5,580,556 12/1996 Horrobin ................................ 424/85.4 73 Assignee: Neuromedica, Inc., Conshohocken, Pa. FOREIGN PATENT DOCUMENTS 30009 6/1981 European Pat. Off.. 21 Appl. No.: 08/462,820 009 1694 10/1983 European Pat. Off.. 22 Filed: Jun. 5, 1995 09 1694 10/1983 European Pat. Off.. 91694 10/1983 European Pat. Off.. Related U.S. Application Data 59-025327 2/1984 Japan. 1153629 6/1989 Japan. 63 Continuation of application No. 08/080,675, Jun. 21, 1993, 1203331 8/1989 Japan. abandoned, which is a continuation of application No. 07/952,191, Sep. 28, 1992, abandoned, which is a continu- (List continued on next page.) ation of application No. 07/577,329, Sep. 4, 1990, aban doned, which is a continuation-in-part of application No. OTHER PUBLICATIONS 07/535,812,tion of application Jun. 11, No. 1990, 67,375.734 abandoned, Eb3-1685, which is a continu-PEN. T. Higuchi et al. 66 Prodrugs as Noye Drug Delivery Sys 4,933,324, which is a continuation-in-part of application No. -

Un Novedoso Enfoque Para El Diseño De Fármacos Antimicrobianos Asistido Por Computadora
TOMOCOMD-CARDD: Un Novedoso Enfoque para el Diseño de Fármacos Antimicrobianos Asistido por Computadora Autora: Yasnay Valdés Rodríguez. Tutores: Prof. Dr. Yovani Marrero Ponce. Prof. MSc. Ricardo Medina Marrero. 2005-2006 La ignorancia afirma o niega rotundamente; la ciencia duda… Voltaire (1694-1778) Quiero dedicar este trabajo a todas aquellas personas que me aprecian y desean lo mejor para mi, especialmente a mis padres. A mi padre Dedico este trabajo con mucho amor, por hacerme comprender que siempre se puede llegar mas lejos, y que no hay nada imposible, solamente hay que luchar... A mi madre Por su infinita bondad, por su sacrificio inigualable. A mis familiares Por todo su apoyo y ayuda que me han mostrado incondicionalmente. A mi hermano Por ser mi fuente de inspiración. A la humanidad “...porque si supiera que el mundo se acaba mañana, yo, hoy todavía, plantaría un árbol” Quiero agradecer a todas aquellas personas que me han ayudado a realizar este sueño: A mis padres por todo el sacrificio realizado, y aún parecerles poco, los amo mucho. A mi madre por estar siempre a mi lado en los buenos y malos momentos ayudándome a levantarme en cualquier recaída. A mi padre por guiarme en la vida y brindarme sus consejos siempre útiles, por darme fuerza y vitalidad. A mi mayor tesoro, mi hermano, que me alumbra de esperanza día a día. A mis tías y primos que me ayudaron mucho, aún estando lejos. A mi novio que me apoyo en todas mis decisiones y con paciencia supo ayudarme. A mis tutores y cotutores que siempre me dieron la mano; especialmente a Yovani por su paciencia, a quien debo gran parte de mi formación como profesional por sus exigencias. -

Identification of a Series of Hair-Cell MET Channel Blockers That Protect Against Aminoglycoside-Induced Ototoxicity
Identification of a series of hair-cell MET channel blockers that protect against aminoglycoside-induced ototoxicity Emma J. Kenyon, … , Corné J. Kros, Guy P. Richardson JCI Insight. 2021;6(7):e145704. https://doi.org/10.1172/jci.insight.145704. Research Article Neuroscience Therapeutics Graphical abstract Find the latest version: https://jci.me/145704/pdf RESEARCH ARTICLE Identification of a series of hair-cell MET channel blockers that protect against aminoglycoside-induced ototoxicity Emma J. Kenyon,1 Nerissa K. Kirkwood,1 Siân R. Kitcher,1 Richard J. Goodyear,1 Marco Derudas,2 Daire M. Cantillon,3 Sarah Baxendale,4 Antonio de la Vega de León,5 Virginia N. Mahieu,1 Richard T. Osgood,1 Charlotte Donald Wilson,1 James C. Bull,6 Simon J. Waddell,3 Tanya T. Whitfield,4 Simon E. Ward,7 Corné J. Kros,1 and Guy P. Richardson1 1Sussex Neuroscience and 2Sussex Drug Discovery Centre, School of Life Sciences, and 3Global Health and Infection, Brighton and Sussex Medical School, University of Sussex, Brighton, United Kingdom. 4Bateson Centre and Department of Biomedical Science, and 5Information School, University of Sheffield, Sheffield, United Kingdom. 6Department of Biosciences, College of Science, Swansea University, Swansea, United Kingdom. 7Medicines Discovery Institute, Cardiff University, Cardiff, United Kingdom. To identify small molecules that shield mammalian sensory hair cells from the ototoxic side effects of aminoglycoside antibiotics, 10,240 compounds were initially screened in zebrafish larvae, selecting for those that protected lateral-line hair cells against neomycin and gentamicin. When the 64 hits from this screen were retested in mouse cochlear cultures, 8 protected outer hair cells (OHCs) from gentamicin in vitro without causing hair-bundle damage. -

Ep 2540295 A1
(19) TZZ Z _T (11) EP 2 540 295 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 02.01.2013 Bulletin 2013/01 A61K 31/404 (2006.01) A61P 25/00 (2006.01) A61P 25/28 (2006.01) (21) Application number: 11171532.2 (22) Date of filing: 27.06.2011 (84) Designated Contracting States: (72) Inventors: AL AT BE BG CH CY CZ DE DK EE ES FI FR GB • Briault, Sylvain GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO 45000 ORLEANS (FR) PL PT RO RS SE SI SK SM TR • Perche, Olivier Designated Extension States: 45380 LA CHAPELLE SAINT MESMIN (FR) BA ME (74) Representative: Thomas, Dean et al (71) Applicants: Cabinet Ores • Centre national de la recherche scientifique 36 rue de St Pétersbourg 75016 Paris (FR) 75008 Paris (FR) • Centre Hospitalier Régional d’Orléans 45032 Orléans Cédex 1 (FR) (54) Compositions for the treatment of Fragile X syndrome (57) The present invention relates to a composition a fluoro-oxindole or a chloro- oxindole for use in the treat- comprising a maxi-K potassium channel opener the use ment of fragile X syndrome. in the treatment of fragile X syndrome. More specifically the present invention relates to a composition comprising EP 2 540 295 A1 Printed by Jouve, 75001 PARIS (FR) 1 EP 2 540 295 A1 2 Description ularly shyness, limited eye contact, memory problems and difficulty with facial encoding and recognition. Many [0001] The present invention relates to compositions individuals with FXS also meet the diagnostic criteria for for the alleviation of neuropsychiatric symptoms and in autism. -

Systematic Evidence Review from the Blood Pressure Expert Panel, 2013
Managing Blood Pressure in Adults Systematic Evidence Review From the Blood Pressure Expert Panel, 2013 Contents Foreword ............................................................................................................................................ vi Blood Pressure Expert Panel ..............................................................................................................vii Section 1: Background and Description of the NHLBI Cardiovascular Risk Reduction Project ............ 1 A. Background .............................................................................................................................. 1 Section 2: Process and Methods Overview ......................................................................................... 3 A. Evidence-Based Approach ....................................................................................................... 3 i. Overview of the Evidence-Based Methodology ................................................................. 3 ii. System for Grading the Body of Evidence ......................................................................... 4 iii. Peer-Review Process ....................................................................................................... 5 B. Critical Question–Based Approach ........................................................................................... 5 i. How the Questions Were Selected ................................................................................... 5 ii. Rationale for the Questions -

O'neill2020 Redacted.Pdf (7.617Mb)
This thesis has been submitted in fulfilment of the requirements for a postgraduate degree (e.g. PhD, MPhil, DClinPsychol) at the University of Edinburgh. Please note the following terms and conditions of use: This work is protected by copyright and other intellectual property rights, which are retained by the thesis author, unless otherwise stated. A copy can be downloaded for personal non-commercial research or study, without prior permission or charge. This thesis cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author. The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author. When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given. Functional Characterisation of Spontaneously Active GABAA Receptors in Rat Dentate Gyrus Granule Cells Nathanael O’Neill B.Medsc. (Hons) Doctor of Philosophy The University of Edinburgh 2020 ii Abstract GABAA receptors (GABAARs) are the principal inhibitory neurotransmitter receptors in the adult mammalian central nervous system. GABAARs mediate two forms of inhibition: fast, phasic conductance; and slow, tonic conductance. Tonic conductance arises due to the persistent activation of GABAARs. This persistent activation can occur by GABA-dependent or GABA- independent mechanisms. Low concentrations of ambient GABA activate high affinity GABAARs located outside the synapse – at peri-/extra-synaptic sites – to generate GABA-dependent tonic conductance. In contrast, GABA-independent tonic conductance is generated by GABAARs that activate spontaneously, in the absence of GABA, due to constitutive receptor gating. -

The Gabaa Receptor 8 Subunit Gene Promoter Fragments to Direct Long - Term Neuron - Specific Expression
THE GABAa RECEPTOR 8 SUBUNIT GENE PROMOTER : CHARACTERISATION AND USE Alexa Brett Roberts Division of Molecular Genetics Institute of Biomedical and Life Sciences University of Glasgow Glasgow A dissertation submitted for the degree of Doctor of Philosophy of the University of Glasgow February 1998 ProQuest Number: 13818612 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest ProQuest 13818612 Published by ProQuest LLC(2018). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLC. ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106- 1346 GIASGOW UNIVERSITY LIBRARY 11110 (coh O f GLASGOW 1 IUNIVERSITT I [LQgAW I For my husband Stuart and for my children Ross and Ailie with love Also to JR (Grandad) in loving memory Preface The work presented in this thesis was performed entirely by the author, except where acknowledged. I declare that my thesis contains unique work and will not be submitted for any other degree, diploma or qualification at any other University. A. Brett Roberts December 1997 Acknowledgements I would like to thank Professor R.W. Davies for giving me the opportunity to study for this degree and for his help throughout the last three years. -

Identification of a Novel Series of Hair-Cell MET Channel Blockers That Protect Against Aminoglycoside-Induced Ototoxicity
Identification of a novel series of hair-cell MET channel blockers that protect against aminoglycoside-induced ototoxicity Emma J. Kenyon, … , Corné J. Kros, Guy P. Richardson. JCI Insight. 2021. https://doi.org/10.1172/jci.insight.145704. Research In-Press Preview Neuroscience Therapeutics Graphical abstract Find the latest version: https://jci.me/145704/pdf Identification of a series of hair-cell MET channel blockers that protect against aminoglycoside-induced ototoxicity Emma J Kenyon1*, Nerissa K Kirkwood1*, Siân R Kitcher1*, Richard J Goodyear1, Marco Derudas2, Daire M Cantillon3, Sarah Baxendale4, Antonio de la Vega de León5 , Virginia N Mahieu1, Richard T Osgood1, Charlotte Donald Wilson1, James C Bull6, Simon J Waddell3, Tanya T Whitfield4, Simon E Ward7, Corné J Kros1 and Guy P Richardson1. 1Sussex Neuroscience, School of Life Sciences, University of Sussex, Brighton, BN1 9QG, UK 2Sussex Drug Discovery Centre, School of Life Sciences, University of Sussex, Brighton, BN1 9QG, UK 3Global Health and Infection, Brighton and Sussex Medical School, University of Sussex, Brighton, BN1 9PX, UK 4Bateson Centre and Department of Biomedical Science, University of Sheffield, Sheffield, S10 2TN, UK 5Information School, University of Sheffield, Regent Court, 211 Portobello, Sheffield. S1 4DP, UK 6Department of Biosciences, College of Science, Swansea University, Swansea, SA2 8PP, UK 7Medicines Discovery Institute, Cardiff University, Cardiff, CF10 3AT, UK *These authors contributed equally to the work and are listed in alphabetical order. Corresponding authors Prof. Guy P Richardson, School of Life Sciences, University of Sussex, Falmer, Brighton, BN1 9QG, UK. Phone 0044 1273 678717. Email: [email protected] Prof. Corné J Kros, School of Life Sciences, University of Sussex, Falmer, Brighton, BN1 9QG, UK.