(12) Patent Application Publication (10) Pub. No.: US 2010/014.3507 A1 Gant Et Al
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Considerations in Perioperative Assessment of Valproic Acid Coagulopathy Claude Abdallah George Washington University
Himmelfarb Health Sciences Library, The George Washington University Health Sciences Research Commons Anesthesiology and Critical Care Medicine Faculty Anesthesiology and Critical Care Medicine Publications 1-2014 Considerations in perioperative assessment of valproic acid coagulopathy Claude Abdallah George Washington University Follow this and additional works at: http://hsrc.himmelfarb.gwu.edu/smhs_anesth_facpubs Part of the Anesthesia and Analgesia Commons APA Citation Abdallah, C. (2014). Considerations in perioperative assessment of valproic acid coagulopathy. Journal of Anaesthesiology Clinical Pharmacology, Volume 30, Issue 1 (). http://dx.doi.org/10.4103/0970-9185.125685 This Journal Article is brought to you for free and open access by the Anesthesiology and Critical Care Medicine at Health Sciences Research Commons. It has been accepted for inclusion in Anesthesiology and Critical Care Medicine Faculty Publications by an authorized administrator of Health Sciences Research Commons. For more information, please contact [email protected]. [Downloaded free from http://www.joacp.org on Tuesday, February 25, 2014, IP: 128.164.86.61] || Click here to download free Android application for this journal Revv iew Article Considerations in perioperative assessment of valproic acid coagulopathy Claude Abdallah Department of Anesthesiology, Children’s National Medical Center, The George Washington University Medical Center, NW Washington, DC, USA Abstract Valproic acid (VPA) is one of the widely prescribed antiepileptic drugs in children with multiple indications. VPA-induced coagulopathy may occur and constitute a pharmacological and practical challenge affecting pre-operative evaluation and management of patients receiving VPA therapy. This review summarizes the different studies documenting the incidence, severity and available recommendations related to this adverse effect. -

Pindolol of the Activation of Postsynaptic 5-HT1A Receptors
Potentiation by (-)Pindolol of the Activation of Postsynaptic 5-HT1A Receptors Induced by Venlafaxine Jean-Claude Béïque, Ph.D., Pierre Blier, M.D., Ph.D., Claude de Montigny, M.D., Ph.D., and Guy Debonnel, M.D. The increase of extracellular 5-HT in brain terminal regions antagonist WAY 100635 (100 g/kg, i.v.). A short-term produced by the acute administration of 5-HT reuptake treatment with VLX (20 mg/kg/day ϫ 2 days) resulted in a inhibitors (SSRI’s) is hampered by the activation of ca. 90% suppression of the firing activity of 5-HT neurons somatodendritic 5-HT1A autoreceptors in the raphe nuclei. in the dorsal raphe nucleus. This was prevented by the The present in vivo electrophysiological studies were coadministration of (-)pindolol (15 mg/kg/day ϫ 2 days). undertaken, in the rat, to assess the effects of the Taken together, these results indicate that (-)pindolol coadministration of venlafaxine, a dual 5-HT/NE reuptake potentiated the activation of postsynaptic 5-HT1A receptors inhibitor, and (-)pindolol on pre- and postsynaptic 5-HT1A resulting from 5-HT reuptake inhibition probably by receptor function. The acute administration of venlafaxine blocking the somatodendritic 5-HT1A autoreceptor, but not and of the SSRI paroxetine (5 mg/kg, i.v.) induced a its postsynaptic congener. These results support and extend suppression of the firing activity of dorsal hippocampus CA3 previous findings providing a biological substratum for the pyramidal neurons. This effect of venlafaxine was markedly efficacy of pindolol as an accelerating strategy in major potentiated by a pretreatment with (-)pindolol (15 mg/kg, depression. -
Therapeutic Class Brand Name P a Status Generic
P A Therapeutic Class Brand Name Status Generic Name Strength Form Absorbable Sulfonamides AZULFIDINE SULFASALAZINE 250MG/5ML ORAL SUSP Absorbable Sulfonamides AZULFIDINE SULFASALAZINE 500MG TABLET Absorbable Sulfonamides AZULFIDINE SULFASALAZINE 500MG TABLET DR Absorbable Sulfonamides BACTRIM DS SULFAMETHOXAZOLE/TRIMETHO 800-160MG TABLET Absorbable Sulfonamides GANTRISIN SULFISOXAZOLE 500MG TABLET Absorbable Sulfonamides GANTRISIN SULFISOXAZOLE ACETYL 500MG/5ML ORAL SUSP Absorbable Sulfonamides GANTRISIN SULFISOXAZOLE ACETYL 500MG/5ML SYRUP Absorbable Sulfonamides SEPTRA SULFAMETHOXAZOLE/TRIMETHO 200-40MG/5 ORAL SUSP Absorbable Sulfonamides SEPTRA SULFAMETHOXAZOLE/TRIMETHO 400-80MG TABLET Absorbable Sulfonamides SULFADIAZINE SULFADIAZINE 500MG TABLET ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 10-20MG CAPSULE ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 2.5-10MG CAPSULE ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 5-10MG CAPSULE ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 5-20MG CAPSULE P A Therapeutic Class Brand Name Status Generic Name Strength Form ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 5-40MG CAPSULE ACE Inhibitor/Calcium Channel Blocker Combination LOTREL AMLODIPINE BESYLATE/BENAZ 10-40MG CAPSULE Acne Agents, Systemic ACCUTANE ISOTRETINOIN 10MG CAPSULE Acne Agents, Systemic ACCUTANE ISOTRETINOIN 20MG CAPSULE Acne Agents, Systemic ACCUTANE -

In Silico Methods for Drug Repositioning and Drug-Drug Interaction Prediction
In silico Methods for Drug Repositioning and Drug-Drug Interaction Prediction Pathima Nusrath Hameed ORCID: 0000-0002-8118-9823 Submitted in total fulfilment of the requirements for the degree of Doctor of Philosophy Department of Mechanical Engineering THE UNIVERSITY OF MELBOURNE May 2018 Copyright © 2018 Pathima Nusrath Hameed All rights reserved. No part of the publication may be reproduced in any form by print, photoprint, microfilm or any other means without written permission from the author. Abstract Drug repositioning and drug-drug interaction (DDI) prediction are two fundamental ap- plications having a large impact on drug development and clinical care. Drug reposi- tioning aims to identify new uses for existing drugs. Moreover, understanding harmful DDIs is essential to enhance the effects of clinical care. Exploring both therapeutic uses and adverse effects of drugs or a pair of drugs have significant benefits in pharmacology. The use of computational methods to support drug repositioning and DDI prediction en- able improvements in the speed of drug development compared to in vivo and in vitro methods. This thesis investigates the consequences of employing a representative training sam- ple in achieving better performance for DDI classification. The Positive-Unlabeled Learn- ing method introduced in this thesis aims to employ representative positives as well as reliable negatives to train the binary classifier for inferring potential DDIs. Moreover, it explores the importance of a finer-grained similarity metric to represent the pairwise drug similarities. Drug repositioning can be approached by new indication detection. In this study, Anatomical Therapeutic Chemical (ATC) classification is used as the primary source to determine the indications/therapeutic uses of drugs for drug repositioning. -
Lab Standard Operating Procedures (Sops)
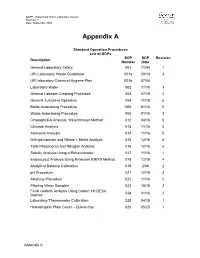
QAPP –Watershed Watch Laboratory Assays Revision: 7 Date: September 2020 Appendix A Standard Operation Procedures List of SOPs SOP SOP Revision Description Number Date General Laboratory Safety 001 11/04 1 URI Laboratory Waste Guidebook 001a 09/13 3 URI laboratory Chemical Hygiene Plan 001b 07/04 Laboratory Water 002 11/16 3 General Labware Cleaning Procedure 003 07/19 4 General Autoclave Operation 004 10/18 6 Bottle Autoclaving Procedure 005 01/16 5 Waste Autoclaving Procedure 006 01/16 3 Chlorophyll-A Analysis, Welschmeyer Method 012 04/18 6 Chloride Analysis 013 11/16 5 Ammonia Analysis 014 11/16 5 Orthophosphate and Nitrate + Nitrite Analysis 015 12/16 5 Total Phosphorus and Nitrogen Analysis 016 12/16 5 Salinity Analysis Using a Refractometer 017 11/16 1 Enterococci Analysis Using Enterolert IDEXX Method 018 12/19 4 Analytical Balance Calibration 019 2/06 2 pH Procedure 021 12/19 3 Alkalinity Procedure 022 11/16 2 Filtering Water Samples 023 10/18 2 Fecal coliform Analysis Using Colilert 18 IDEXX 024 01/16 2 Method Laboratory Thermometer Calibration 025 04/19 1 Heterotrophic Plate Count – Quanti-tray 026 05/20 1 Appendix A Standard Operating Procedure 001 Date: 11/04 General Laboratory Safety Revision: 1 Author: Linda Green University of Rhode Island Watershed Watch 1.0 PURPOSE AND DESCRIPTION LAB SAFETY IS EVERYBODY’S JOB! Please be sure to familiarize yourself with these general procedures, as well as the specific handling requirements included in the Standard Operating Procedure (SOP) for each analysis/process. Further general information regarding University of Rhode Island standards for health and safety are found in SOP 001a – University Safety and Waste Handling Document. -

AHFS Pharmacologic-Therapeutic Classification System
AHFS Pharmacologic-Therapeutic Classification System Abacavir 48:24 - Mucolytic Agents - 382638 8:18.08.20 - HIV Nucleoside and Nucleotide Reverse Acitretin 84:92 - Skin and Mucous Membrane Agents, Abaloparatide 68:24.08 - Parathyroid Agents - 317036 Aclidinium Abatacept 12:08.08 - Antimuscarinics/Antispasmodics - 313022 92:36 - Disease-modifying Antirheumatic Drugs - Acrivastine 92:20 - Immunomodulatory Agents - 306003 4:08 - Second Generation Antihistamines - 394040 Abciximab 48:04.08 - Second Generation Antihistamines - 394040 20:12.18 - Platelet-aggregation Inhibitors - 395014 Acyclovir Abemaciclib 8:18.32 - Nucleosides and Nucleotides - 381045 10:00 - Antineoplastic Agents - 317058 84:04.06 - Antivirals - 381036 Abiraterone Adalimumab; -adaz 10:00 - Antineoplastic Agents - 311027 92:36 - Disease-modifying Antirheumatic Drugs - AbobotulinumtoxinA 56:92 - GI Drugs, Miscellaneous - 302046 92:20 - Immunomodulatory Agents - 302046 92:92 - Other Miscellaneous Therapeutic Agents - 12:20.92 - Skeletal Muscle Relaxants, Miscellaneous - Adapalene 84:92 - Skin and Mucous Membrane Agents, Acalabrutinib 10:00 - Antineoplastic Agents - 317059 Adefovir Acamprosate 8:18.32 - Nucleosides and Nucleotides - 302036 28:92 - Central Nervous System Agents, Adenosine 24:04.04.24 - Class IV Antiarrhythmics - 304010 Acarbose Adenovirus Vaccine Live Oral 68:20.02 - alpha-Glucosidase Inhibitors - 396015 80:12 - Vaccines - 315016 Acebutolol Ado-Trastuzumab 24:24 - beta-Adrenergic Blocking Agents - 387003 10:00 - Antineoplastic Agents - 313041 12:16.08.08 - Selective -

Therapeutic Class Overview Anticonvulsants
Therapeutic Class Overview Anticonvulsants INTRODUCTION Epilepsy is a disease of the brain defined by any of the following (Fisher et al 2014): ○ At least 2 unprovoked (or reflex) seizures occurring > 24 hours apart; ○ 1 unprovoked (or reflex) seizure and a probability of further seizures similar to the general recurrence risk (at least 60%) after 2 unprovoked seizures, occurring over the next 10 years; ○ Diagnosis of an epilepsy syndrome. Types of seizures include generalized seizures, focal (partial) seizures, and status epilepticus (Centers for Disease Control and Prevention [CDC] 2018, Epilepsy Foundation 2016). ○ Generalized seizures affect both sides of the brain and include: . Tonic-clonic (grand mal): begin with stiffening of the limbs, followed by jerking of the limbs and face . Myoclonic: characterized by rapid, brief contractions of body muscles, usually on both sides of the body at the same time . Atonic: characterized by abrupt loss of muscle tone; they are also called drop attacks or akinetic seizures and can result in injury due to falls . Absence (petit mal): characterized by brief lapses of awareness, sometimes with staring, that begin and end abruptly; they are more common in children than adults and may be accompanied by brief myoclonic jerking of the eyelids or facial muscles, a loss of muscle tone, or automatisms. ○ Focal seizures are located in just 1 area of the brain and include: . Simple: affect a small part of the brain; can affect movement, sensations, and emotion, without a loss of consciousness . Complex: affect a larger area of the brain than simple focal seizures and the patient loses awareness; episodes typically begin with a blank stare, followed by chewing movements, picking at or fumbling with clothing, mumbling, and performing repeated unorganized movements or wandering; they may also be called “temporal lobe epilepsy” or “psychomotor epilepsy” . -

Pharmacokinetic Drug–Drug Interactions Among Antiepileptic Drugs, Including CBD, Drugs Used to Treat COVID-19 and Nutrients
International Journal of Molecular Sciences Review Pharmacokinetic Drug–Drug Interactions among Antiepileptic Drugs, Including CBD, Drugs Used to Treat COVID-19 and Nutrients Marta Kara´zniewicz-Łada 1 , Anna K. Główka 2 , Aniceta A. Mikulska 1 and Franciszek K. Główka 1,* 1 Department of Physical Pharmacy and Pharmacokinetics, Poznan University of Medical Sciences, 60-781 Pozna´n,Poland; [email protected] (M.K.-Ł.); [email protected] (A.A.M.) 2 Department of Bromatology, Poznan University of Medical Sciences, 60-354 Pozna´n,Poland; [email protected] * Correspondence: [email protected]; Tel.: +48-(0)61-854-64-37 Abstract: Anti-epileptic drugs (AEDs) are an important group of drugs of several generations, rang- ing from the oldest phenobarbital (1912) to the most recent cenobamate (2019). Cannabidiol (CBD) is increasingly used to treat epilepsy. The outbreak of the SARS-CoV-2 pandemic in 2019 created new challenges in the effective treatment of epilepsy in COVID-19 patients. The purpose of this review is to present data from the last few years on drug–drug interactions among of AEDs, as well as AEDs with other drugs, nutrients and food. Literature data was collected mainly in PubMed, as well as google base. The most important pharmacokinetic parameters of the chosen 29 AEDs, mechanism of action and clinical application, as well as their biotransformation, are presented. We pay a special attention to the new potential interactions of the applied first-generation AEDs (carba- Citation: Kara´zniewicz-Łada,M.; mazepine, oxcarbazepine, phenytoin, phenobarbital and primidone), on decreased concentration Główka, A.K.; Mikulska, A.A.; of some medications (atazanavir and remdesivir), or their compositions (darunavir/cobicistat and Główka, F.K. -

Classification of Medicinal Drugs and Driving: Co-Ordination and Synthesis Report
Project No. TREN-05-FP6TR-S07.61320-518404-DRUID DRUID Driving under the Influence of Drugs, Alcohol and Medicines Integrated Project 1.6. Sustainable Development, Global Change and Ecosystem 1.6.2: Sustainable Surface Transport 6th Framework Programme Deliverable 4.4.1 Classification of medicinal drugs and driving: Co-ordination and synthesis report. Due date of deliverable: 21.07.2011 Actual submission date: 21.07.2011 Revision date: 21.07.2011 Start date of project: 15.10.2006 Duration: 48 months Organisation name of lead contractor for this deliverable: UVA Revision 0.0 Project co-funded by the European Commission within the Sixth Framework Programme (2002-2006) Dissemination Level PU Public PP Restricted to other programme participants (including the Commission x Services) RE Restricted to a group specified by the consortium (including the Commission Services) CO Confidential, only for members of the consortium (including the Commission Services) DRUID 6th Framework Programme Deliverable D.4.4.1 Classification of medicinal drugs and driving: Co-ordination and synthesis report. Page 1 of 243 Classification of medicinal drugs and driving: Co-ordination and synthesis report. Authors Trinidad Gómez-Talegón, Inmaculada Fierro, M. Carmen Del Río, F. Javier Álvarez (UVa, University of Valladolid, Spain) Partners - Silvia Ravera, Susana Monteiro, Han de Gier (RUGPha, University of Groningen, the Netherlands) - Gertrude Van der Linden, Sara-Ann Legrand, Kristof Pil, Alain Verstraete (UGent, Ghent University, Belgium) - Michel Mallaret, Charles Mercier-Guyon, Isabelle Mercier-Guyon (UGren, University of Grenoble, Centre Regional de Pharmacovigilance, France) - Katerina Touliou (CERT-HIT, Centre for Research and Technology Hellas, Greece) - Michael Hei βing (BASt, Bundesanstalt für Straßenwesen, Germany). -

1-(4-Amino-Cyclohexyl)
(19) & (11) EP 1 598 339 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C07D 211/04 (2006.01) C07D 211/06 (2006.01) 24.06.2009 Bulletin 2009/26 C07D 235/24 (2006.01) C07D 413/04 (2006.01) C07D 235/26 (2006.01) C07D 401/04 (2006.01) (2006.01) (2006.01) (21) Application number: 05014116.7 C07D 401/06 C07D 403/04 C07D 403/06 (2006.01) A61K 31/44 (2006.01) A61K 31/48 (2006.01) A61K 31/415 (2006.01) (22) Date of filing: 18.04.2002 A61K 31/445 (2006.01) A61P 25/04 (2006.01) (54) 1-(4-AMINO-CYCLOHEXYL)-1,3-DIHYDRO-2H-BENZIMIDAZOLE-2-ONE DERIVATIVES AND RELATED COMPOUNDS AS NOCICEPTIN ANALOGS AND ORL1 LIGANDS FOR THE TREATMENT OF PAIN 1-(4-AMINO-CYCLOHEXYL)-1,3-DIHYDRO-2H-BENZIMIDAZOLE-2-ON DERIVATE UND VERWANDTE VERBINDUNGEN ALS NOCICEPTIN ANALOGE UND ORL1 LIGANDEN ZUR BEHANDLUNG VON SCHMERZ DERIVÉS DE LA 1-(4-AMINO-CYCLOHEXYL)-1,3-DIHYDRO-2H-BENZIMIDAZOLE-2-ONE ET COMPOSÉS SIMILAIRES POUR L’UTILISATION COMME ANALOGUES DU NOCICEPTIN ET LIGANDES DU ORL1 POUR LE TRAITEMENT DE LA DOULEUR (84) Designated Contracting States: • Victory, Sam AT BE CH CY DE DK ES FI FR GB GR IE IT LI LU Oak Ridge, NC 27310 (US) MC NL PT SE TR • Whitehead, John Designated Extension States: Newtown, PA 18940 (US) AL LT LV MK RO SI (74) Representative: Maiwald, Walter (30) Priority: 18.04.2001 US 284666 P Maiwald Patentanwalts GmbH 18.04.2001 US 284667 P Elisenhof 18.04.2001 US 284668 P Elisenstrasse 3 18.04.2001 US 284669 P 80335 München (DE) (43) Date of publication of application: (56) References cited: 23.11.2005 Bulletin 2005/47 EP-A- 0 636 614 EP-A- 0 990 653 EP-A- 1 142 587 WO-A-00/06545 (62) Document number(s) of the earlier application(s) in WO-A-00/08013 WO-A-01/05770 accordance with Art. -

Properties and Units in Clinical Pharmacology and Toxicology
Pure Appl. Chem., Vol. 72, No. 3, pp. 479–552, 2000. © 2000 IUPAC INTERNATIONAL FEDERATION OF CLINICAL CHEMISTRY AND LABORATORY MEDICINE SCIENTIFIC DIVISION COMMITTEE ON NOMENCLATURE, PROPERTIES, AND UNITS (C-NPU)# and INTERNATIONAL UNION OF PURE AND APPLIED CHEMISTRY CHEMISTRY AND HUMAN HEALTH DIVISION CLINICAL CHEMISTRY SECTION COMMISSION ON NOMENCLATURE, PROPERTIES, AND UNITS (C-NPU)§ PROPERTIES AND UNITS IN THE CLINICAL LABORATORY SCIENCES PART XII. PROPERTIES AND UNITS IN CLINICAL PHARMACOLOGY AND TOXICOLOGY (Technical Report) (IFCC–IUPAC 1999) Prepared for publication by HENRIK OLESEN1, DAVID COWAN2, RAFAEL DE LA TORRE3 , IVAN BRUUNSHUUS1, MORTEN ROHDE1, and DESMOND KENNY4 1Office of Laboratory Informatics, Copenhagen University Hospital (Rigshospitalet), Copenhagen, Denmark; 2Drug Control Centre, London University, King’s College, London, UK; 3IMIM, Dr. Aiguader 80, Barcelona, Spain; 4Dept. of Clinical Biochemistry, Our Lady’s Hospital for Sick Children, Crumlin, Dublin 12, Ireland #§The combined Memberships of the Committee and the Commission (C-NPU) during the preparation of this report (1994–1996) were as follows: Chairman: H. Olesen (Denmark, 1989–1995); D. Kenny (Ireland, 1996); Members: X. Fuentes-Arderiu (Spain, 1991–1997); J. G. Hill (Canada, 1987–1997); D. Kenny (Ireland, 1994–1997); H. Olesen (Denmark, 1985–1995); P. L. Storring (UK, 1989–1995); P. Soares de Araujo (Brazil, 1994–1997); R. Dybkær (Denmark, 1996–1997); C. McDonald (USA, 1996–1997). Please forward comments to: H. Olesen, Office of Laboratory Informatics 76-6-1, Copenhagen University Hospital (Rigshospitalet), 9 Blegdamsvej, DK-2100 Copenhagen, Denmark. E-mail: [email protected] Republication or reproduction of this report or its storage and/or dissemination by electronic means is permitted without the need for formal IUPAC permission on condition that an acknowledgment, with full reference to the source, along with use of the copyright symbol ©, the name IUPAC, and the year of publication, are prominently visible. -

Clonazepam/Ethosuximide 479 Abnormal Movements, and for the Treatment of Panic BNFC Suggests Giving the Following Doses by Slow Intravenous 3
Clonazepam/Ethosuximide 479 abnormal movements, and for the treatment of panic BNFC suggests giving the following doses by slow intravenous 3. Peled R, Lavie P. Double-blind evaluation of clonazepam on pe- injection over at least 2 minutes according to age: riodic leg movements in sleep. J Neurol Neurosurg Psychiatry disorder (see Psychiatric Disorders, below). 1987; 50: 1679–81. For epilepsy and myoclonus treatment is started with • neonates: 100 micrograms/kg, repeated if necessary after 24 4. Saletu M, et al. Restless legs syndrome (RLS) and periodic limb hours movement disorder (PLMD): acute placebo-controlled sleep lab- small doses that are progressively increased to an opti- oratory studies with clonazepam. Eur Neuropsychopharmacol • 1 month to 12 years: 50 micrograms/kg (maximum 1 mg), re- 2001; 11: 153–61. mum dose according to response. Total daily doses peated if necessary may initially be taken in 3 or 4 divided doses; however, 5. Saletu A, et al. On the pharmacotherapy of sleep bruxism: place- Older children may be given the usual adult dose. bo-controlled polysomnographic and psychometric studies with once the maintenance dose has been reached, the daily clonazepam. Neuropsychobiology 2005; 51: 214–25. In children aged over 1 month, these doses by injection may be amount may be given as a single dose at night. In the followed by an intravenous infusion of 10 micrograms/kg per Stiff-man syndrome. Clonazepam has been used as an alter- UK the initial oral dose is 1 mg (500 micrograms in the native to diazepam in the management of stiff-man syndrome hour, adjusted according to response to a maximum of 1 elderly) at night for 4 nights gradually increased over 2 60 micrograms/kg per hour.