Carbothermic Production of Hexagonal Boron Nitride A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Equations of State of New Boron-Rich Selenides B6se and B12se
Equations of state of new boron-rich selenides B6Se and B12Se Kirill A. Cherednichenko,1,2 Yann Le Godec,3 and Vladimir L. Solozhenko 1,* 1 LSPM–CNRS, Université Sorbonne Paris Nord, 93430 Villetaneuse, France 2 Department of Physical and Colloid Chemistry, Gubkin University, Moscow, 119991, Russia 3 IMPMC–CNRS, UPMC Sorbonne Universités, 75005 Paris, France Abstract Two novel of boron-rich selenides, orthorhombic B6Se and rhombohedral B12Se, have been recently synthesized at high pressure – high temperature conditions. Room-temperature compressibilities of these phases were studied in a diamond anvil cell using synchrotron powder X-ray diffraction. A fit of experimental p-V data by third-order Birch-Murnaghan equation of state yielded the bulk moduli of 155(2) GPa for B12Se and 144(3) GPa for B6Se. No pressure-induced phase transitions have been observed in the studied pressure range, i.e. up to 35 GPa. Keywords : boron-rich selenides; high pressure; equation of state Introduction Boron-rich compounds containing B12-icosahedra have been known since 1960-s [1]. The majority of these compounds are the structural derivatives of elemental α-rhombohedral boron (α-B12) with general stoichiometry: B12Xy (where X is interstitial atom, y ≤ 2). They received a considerable attention due to the high hardness, chemical inertness, thermal conductivity, radiation resistance and unusual electronic properties [2-5]. For instance, boron carbide B12C3 is hard compound widely used as abrasive and armor material [6,7], boron suboxide B12O2 is the hardest known oxide [8,9], and B12As2 was found to be extremely stable under electron bombardment and able to the self-healing [10]. -

Towards the Theory of Hardness of Materials
Towards the theory of hardness of materials. Artem R. Oganov1,2, Andriy O. Lyakhov1 1 Department of Geosciences, Department of Physics and Astronomy, and New York Center for Computational Sciences, Stony Brook University, Stony Brook, New York 11794-2100, USA 2 Geology Department, Moscow State University, 119992 Moscow, Russia Recent studies showed that hardness, a complex property, can be calculated using very simple approaches or even analytical formulae. These form the basis for evaluating controversial experimental results (as we illustrate for TiO2-cotunnite) and enable a systematic search for novel hard materials, for instance, using global optimization algorithms (as we show on the example of SiO2 polymorphs). We all know that diamond is very hard, while graphite and talc are soft. Hardness, as a property of materials, determines many of their technological applications, but remains a poorly understood property. The aim of this Special Issue is to review some of the most important recent developments in the understanding of hardness. Mohs’s relative scale of hardness appeared in the XIX century and is still widely used by mineralogists; in this scale talc has hardness 1, and diamond has hardness 10. There are several absolute definitions of hardness – the most popular ones being the Knoop and Vickers tests of hardness, which involve indentation (rather than scratching, as in Mohs’s hardness). The absolute hardness is measured in GPa, the same units as pressure or elastic moduli (bulk modulus, shear modulus). This hints that hardness may be correlated with the elastic properties – indeed, there is such a correlation, especially with the shear modulus. -

In Situ High-Pressure Synthesis of New Outstanding Light-Element Materials Under Industrial P-T Range
materials Review In Situ High-Pressure Synthesis of New Outstanding Light-Element Materials under Industrial P-T Range Yann Le Godec 1,* and Alexandre Courac 1,2 1 Institut de Minéralogie, de Physique des Matériaux et de Cosmochimie (IMPMC), Sorbonne Université, UMR CNRS 7590, Muséum National d’Histoire Naturelle, IRD UMR 206, 75005 Paris, France; [email protected] 2 Institut Universitaire de France, IUF, 75005 Paris, France * Correspondence: [email protected] Abstract: High-pressure synthesis (which refers to pressure synthesis in the range of 1 to several GPa) adds a promising additional dimension for exploration of compounds that are inaccessible to traditional chemical methods and can lead to new industrially outstanding materials. It is nowadays a vast exciting field of industrial and academic research opening up new frontiers. In this context, an emerging and important methodology for the rapid exploration of composition-pressure-temperature- time space is the in situ method by synchrotron X-ray diffraction. This review introduces the latest advances of high-pressure devices that are adapted to X-ray diffraction in synchrotrons. It focuses particularly on the “large volume” presses (able to compress the volume above several mm3 to pressure higher than several GPa) designed for in situ exploration and that are suitable for discovering and scaling the stable or metastable compounds under “traditional” industrial pressure range (3–8 GPa). We illustrated the power of such methodology by (i) two classical examples of “reference” superhard high-pressure materials, diamond and cubic boron nitride c-BN; and (ii) recent successful in situ high-pressure syntheses of light-element compounds that allowed expanding Citation: Le Godec, Y.; Courac, A. -

Synthesis of Li2s-Carbon Cathode Materials Via Carbothermic Reduction of Li2so4
ORIGINAL RESEARCH published: 04 June 2019 doi: 10.3389/fenrg.2019.00053 Synthesis of Li2S-Carbon Cathode Materials via Carbothermic Reduction of Li2SO4 Jiayan Shi 1, Jian Zhang 2, Yifan Zhao 2, Zheng Yan 1, Noam Hart 1 and Juchen Guo 1,2* 1 Department of Chemical and Environmental Engineering, University of California, Riverside, Riverside, CA, United States, 2 Materials Science and Engineering Program, University of California, Riverside, Riverside, CA, United States Pre-lithiated sulfur materials are promising cathode for lithium-sulfur batteries. The synthesis of lithium sulfide-carbon (Li2S-C) composite by carbothermic reduction of lithium sulfate (Li2SO4) is investigated in this study. The relationship between reaction temperature and the consumption of carbon in the carbothermic reduction of Li2SO4 is first investigated to precisely control the carbon content in the resultant Li2S-C composites. To understand the relationship between the material structure and the electrochemical properties, Li2S-C composites with the same carbon content are subsequently synthesized by controlling the mass ratio of Li2SO4/carbon and the reaction temperature. Systematic electrochemical analyses and microscopic characterizations Edited by: demonstrate that the size of the Li S particles dispersed in the carbon matrix is the key Wen Liu, 2 Beijing University of Chemical parameter determining the electrochemical performance. A reversible capacity of 600 Technology, China −1 mAh g is achieved under lean electrolyte condition with high Li2S areal loading. -

Carbothermic Reduction Kinetics of Ilmenite Concentrates Catalyzed by Sodium Silicate and Microwave- Absorbing Characteristics O
Available on line at Association of the Chemical Engineers of Serbia AChE www.ache.org.rs/CICEQ Chemical Industry & Chemical Engineering Quarterly 19 (3) 423−433 (2013) CI&CEQ WEI LI1,2 CARBOTHERMIC REDUCTION KINETICS OF JINHUI PENG1 1 ILMENITE CONCENTRATES CATALYZED BY SHENGHUI GUO SODIUM SILICATE AND MICROWAVE- LIBO ZHANG1 GUO CHEN1 ABSORBING CHARACTERISTICS OF HONGYING XIA1 REDUCTIVE PRODUCTS 1 Key Laboratory of Unconventional Carbothermic reduction kinetics of ilmenite concentrates catalyzed by sodium Metallurgy, Ministry of Education, silicate were investigated; the reduction degree of ilmenite concentrates Kunming University of Science and reduction reaction was determined as R = 4/7(16y + 56x)(ΔWΣ - f W)/(16y + Technology, Yunnan, China A-P 2Faculty of Science, Kunming + 56x + 112). The results show that the reaction activation energy of initial University of Science and stage and later stage is 36.45 and 135.14 kJ/mol, respectively. There is a great Technology, Yunnan, China change in the reduction rate at temperatures of 1100 and 1150 °C; the catal- ysis effect and change of reduction rate were evaluated by TG and DSC curves SCIENTIFIC PAPER of sodium silicate. Microwave-absorbing characteristics of reduction products were measured by the method of microwave cavity perturbation. It was found UDC 544.47/.478 that microwave absorbing characteristics of reduction products obtained at ° DOI 10.2298/CICEQ120421077L temperatures of 900, 1100 and 1150 C have significant differences. XRD characterization results explained the formation and accumulation of reduction product Fe, and pronounced changes of microwave absorbing characteristics due to the decrease of the content of ilmenite concentrates. Keywords: sodium silicate; ilmenite concentrates; catalytic reduction; kinetics; microwave absorbing characteristics. -

Some Recent Contributions to Our Knowledge of Metallic Reducing Agents.' by H
SOME RECENT CONTRIBUTIONS TO OUR KNOWLEDGE OF METALLIC REDUCING AGENTS.' BY H. F. KELLER. N the extraction of metals from their oxygen compounds, car- I bon is certainly the most iniportant and useful reducing agent. Its great affinity for oxjigen is utilized in the manufac- ture of iron and steel ; the coiiimercial production of other useful metals, such as copper, lead, zinc, and tin is accomplished, either directly or indirectly, with its aid; and under its influ- ence even the energetic metals of the alkalies release their pow- erful grip upon oxygen. Contrasted with this, the use of other substances for the purpose of abstracting oxygen is almost insig- nificant. Is it surprising, therefore, that carbon is looked upon as the reducing agent par excellence, or that the very idea of reduction seem to us inseparably associated with this element ? Hut the reducing power of a substance depends, in a large meas- ure, upon temperature. It is well known to chemists that, under certain conditions, many metals have affinities which are far more powerful than those of carbon. When we glance over the pages of our chemical text-books, we see described there numer- ous experiments based upon the superior combining power of iron, aluminum, zinc, magnesium, and the alkali metals. The great Swedish chemist who arranged the elements according to their supposed affinities in an electro-chemical series, was among the first to employ the positive alkali metals for isolating and preparing other elements. His method consisted in decom- posing the halogen compounds by means of potassium ; and it was subsequently improved by Berzelius' distinguished disciple , Wohler. -

United States Patent (19) 11 Patent Number: 5,366,526 Ellison-Hayashi Et Al
US005366526A United States Patent (19) 11 Patent Number: 5,366,526 Ellison-Hayashi et al. 45 Date of Patent: Nov. 22, 1994 54 METHOD OF ABRADING WITH BORON tals:', Stanford Research Institute, Menlo Park, Calif., SUBOXIDE (BXO) AND THE BORON Jan., 1963. SUBOXIDE (BXO) ARTICLES AND H. Tracy Hall and Lane A. Compton, "Group IV Ana COMPOST ONUSED logs and High Pressure, High Temperature Synthesis of 75 Inventors: Cristan Ellison-Hayashi, Salt Lake B2O’, Inorg. Chem. 4 (1965) 1213. City, Utah; George T. Emond, W. C. Simmons, "Progress and Planning Report on Southington, Conn.; Shih Y. Kuo, Boron Suboxide B60', Air Force Materials Labora Salt Lake City, Utah tory, Mar., 1968. H. Wrerheit, P. Runow, and H. G. Leis, "On 73) Assignee: Norton Company, Worcester, Mass. Boron-Suboxide Surface Layers and Surface States of 21 Appl. No.: 51,114 B-Rhombohedral Boron:”, Phys. Stat. Sol. (a) 2, K125 (1970). 22 Filed: Apr. 21, 1993 E. V. Zubova, K. P. Burdina, "Synthesis of B6O Under Pressure', Dokl. Akad. Nauk. SSR, 197 (5) (1971) Related U.S. Application Data 1055-1056. 63 Continuation-in-part of Ser. No. 920,357, Jul. 28, 1992, which is a continuation-in-part of Ser. No. 729,467, Jul. (List continued on next page.) 12, 1991, Pat. No. 5,135,892. 51) int. Cl. ................................................ C09K3/14 Primary Examiner-Wayne Langel 52 U.S. C. ......................................... 51/307; 51/309 Attorney, Agent, or Firm-Brian M. Kolkowski 58 Field of Search ............................ 423/278; 501/1; 51/281 R, 307, 309 57 ABSTRACT 56 References Cited A method of removing material from a surface compris U.S. -

CARBOTHERMIC REDUCTION of MECHANICALLY ACTIVATED Nio-CARBON MIXTURE: NON-ISOTHERMAL KINETICS
J. Min. Metall. Sect. B-Metall. 54 (3) B (2018) 313 - 322 Journal of Mining and Metallurgy, Section B: Metallurgy CARBOTHERMIC REDUCTION OF MECHANICALLY ACTIVATED NiO-CARBON MIXTURE: NON-ISOTHERMAL KINETICS S. Bakhshandeh, N. Setoudeh *, M. Ali Askari Zamani, A. Mohassel Yasouj University, Materials Engineering Department, Yasouj, Iran (Received 23 March 2018; accepted 10 December 2018) Abstract The effect of mechanical activation on the carbothermic reduction of nickel oxide was investigated. Mixtures of nickel oxide and activated carbon (99% carbon) were milled for different periods of time in a planetary ball mill. The unmilled mixture and milled samples were subjected to thermogravimetric analysis (TGA) under an argon atmosphere and their solid products of the reduction reaction were studied using XRD experiments. TGA showed that the reduction of NiO started at ~800 and ~720 in un-milled and one-hour milled samples respectively whilst after 25h of milling it decreased to about 430 . The kinetics parameters of carbothermic reduction were determined using non-isothermal method (Coats-Redfern Method) for un-milled and milled samples. The activation energy was determined to be about 222 kJ mol -1 for un-milled mixture whilst it was decreased to about 148 kJ mol -1 in 25-h milled sample. The decrease in the particle size/crystallite size of the℃ milled samp℃les resulted in a significant drop in the reaction temperature. ℃ Keywords : Ball milling; Carbothermic reduction; Nickel oxide; Non-isothermal kinetics; Thermo-gravimetric analysis (TGA). 1. Introduction solid product catalyzes the reaction rate [ 11 ]. It has been shown that mechanical activation by There have been many investigations on the milling reactants together with extended periods of carbothermic reduction of metal oxide in the literature time can intensify the reactions [ 16-19 ]. -

Optimization of the Synthesis of Boron Suboxide Powders
Proceedings of the World Congress on Engineering and Computer Science 2014 Vol II WCECS 2014, 22-24 October, 2014, San Francisco, USA Optimization of the Synthesis of Boron Suboxide Powders Enoch N. Ogunmuyiwa*, Oluwagbenga T. Johnson, Member, IAENG, Iakovos Sigalas, Mathias Hermann and Ayo S. Afolabi, Member, IAENG 5 MPa.m0.5 and significantly better than that of a single Abstract— B6O powders were produced from the reaction crystal of cBN at 2.8 MPa.m0.5 [3]. B6O has a better between boric acid and amorphous boron powders at the thermal stability compared to that of diamond [2, 4] and can o reaction temperatures between 300 and 1400 C for 6 hours. be produced without high pressure [5–8]. All these The powders produced were characterized in terms of particle properties suggest that B O may be a good candidate for size, phase analysis and composition, product yield as well as 6 morphology. Increase in temperature increases both the yield cutting tool and other wear part applications where abrasive as well as the particle size of the produced powders. XRD wear resistance is important. pattern obtained also showed improved crystallinity of the Several publications and patents have reported that B6O produced powder as the temperature increases. SEM image powder can be produced mainly via two techniques: the obtained at higher temperature clearly showed improved reaction of boron with boron oxide (B2O3) at moderately crystallinity (star-like crystals) as the reaction temperature was o high temperatures and pressure, and the reaction of boron increased. The B6O powders synthesised at 1300 C for 6 hours with a metal oxide at temperatures ranging between 1200oC had the optimum yield of over 95%. -
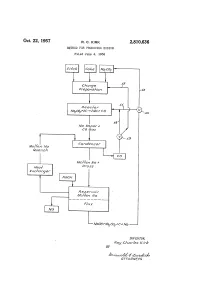
Wooh+Wacof C. Ma
Oct. 22, 1957 R. C. KRK 2,810,636 METHOD FOR PRODUCING SODIUM Filed June 4, 1956 Charge tAeac for Waco, 7-2C-2Wa CO Wa Vooor 7 CO Gas Mo/fern Wa Gornoeraser Guerch Mo/fer? Wa 7. Aea/ O/ro SS Af xcharger Aeser/or Mo/fer? Wa WoOh+WaCOf c. Ma INVENTOR. - Aoy CAar/es kirA BY A77OAAWEYS 2,810,636 United States Patent Office Patented Oct. 22, 1957 1. 2 Some dross is inevitably formed during this condensa tion operation and is separated from the molten sodium 2,810,636 by fluxing with sodium hydroxide. It has been discovered that the carbon monoxide which METHOD FOR PRODUCING SODUM is obtained as a by-product from the reaction of carbon with sodium carbonate will react with the sodium hy Roy.Chemical Charles Company,Kirk, Midland, Midland, Michassignor Mich., a corporation to The Dow of droxide in the flux at temperatures of at least 318 C. Delaware (at which temperature the sodium hydroxide is fluid or molten) to produce sodium carbonate. The thus treated Application June 4, 1956, Serial No. 589,198 0 flux may then be fed into the reactor with added carbon 7 Claims. (C. 75-66) to produce more sodium, since the higher melting sodium carbonate does not attack the graphite lining of the re actor. In this manner a cyclical process is obtained which recovers almost 100 percent of the sodium values present. This invention relates to a process for producing So 5 Since the sodium values in the dross are almost entirely dium and is particularly directed to an improved cyclical recovered, economical modifications of the sodium car method of making sodium by the carbothermic reduction bonate reduction process which previously could not be of sodium carbonate. -

Carbothermic Reduction of Alumina with Carbon in Vacuum
J. Cent. South Univ. (2012) 19: 1813−1816 DOI: 10.1007/s1177101212130 Carbothermic reduction of alumina with carbon in vacuum YU Qingchun(郁青春), YUAN Haibin(袁海滨), ZHU Fulong(朱富龙), ZHANG Han(张晗), WANG Chen(王辰), LIU Dachun(刘大春), YANG Bin(杨斌) National Engineering Laboratory of Vacuum Metallurgy, Key Laboratory of Breeding Base of Complex Nonferrous Metal Resources Clear Utilization in Yunnan Province, State Key Laboratory of Nonferrous Metal Vacuum Metallurgy, Kunming University of Science and Technology, Kunming 650093, China © Central South University Press and SpringerVerlag Berlin Heidelberg 2012 Abstract: Carbothermic reduction alumina in vacuum was conducted, and the products were analysed by means of XRD and gas chromatography. Thermodynamic analysis shows that in vacuum the initial carbothermic reduction reaction temperature reduces compared with that under normal pressure, and the preferential order of products is Al4O4C, Al4C3, Al2OC, Al2O and Al. Experiment results show that the carbothermic reduction products of alumina are Al4O4C and Al4C3, and neither Al2OC, Al2O or Al was found. During the carbothermic reduction process, the reaction rate of Al2O3 and carbon decreases gradually with increasing time. Meanwhile, lower system pressure or higher temperature is beneficial to the carbothermic reduction of alumina process. Al4O4C is firstly formed in the carbothermic reaction, and then Al4C3 is formed in lower system pressure or at higher temperature. Key words: alumina; carbothermic reduction; vacuum; aluminum the formation of aluminum carbide and oxycarbide 1 Introduction byproducts complicates the carbothermic reduction and chlorination process. The thermodynamic study of Aluminum is currently produced industrially via the chemical reactions provides a basic understanding of the HallHe´roult process by dissolving Al2O3 in fused process prior to designing suitable reaction experiments, NaFAlF3 (cryolite) followed by direct current and provides a useful guideline for the selection of electrolysis. -

Boron Suboxide and Boron Subphosphide Crystals: Hard Ceramics That Shear Without Brittle Failure Qi an and William A
Supplementary Information Boron Suboxide and Boron Subphosphide Crystals: Hard Ceramics that Shear without Brittle Failure Qi An and William A. Goddard III *, Materials and Process Simulation Center, California Institute of Technology, Pasadena, CA 91125, U. S. A. *Corresponding author E-mail: [email protected] Table S1. The predicted elastic moduli (GPa) for the boron suboxide crystal framework. The calculated elastic constants used a non-standard set of cell axes. Cijkl XX YY ZZ YZ ZX XY XX 468.9 92.2 75.0 26.2 -16.4 -22.7 YY 92.2 531.5 91.1 -39.6 -6.6 -39.9 ZZ 75.0 91.1 571.1 -20.6 -34.1 -16.3 XY -22.7 -39.9 -16.3 0.0 29.7 216.1 YZ 26.2 -39.6 -20.6 204.3 -7.5 0.0 ZX -16.4 -6.6 -34.1 -7.5 196.5 29.7 Figure S1 Figure S1. The local atomic structures for shearing along the (01Ŭ11)/<101Ŭ1> slip system. (a) the intact structure. (b) the 0.565 strain, at which the structure recovers to the original structure, but with the “c” axis along [111]r direction. (c) the 1.13 strain, before the second recovery of the original structure. (d) the 1.16 strain, where the original structure is recovered the second time, but with the “c” axis along the [101]r direction. The boron and oxygen atoms are in green and red, respectively. Figure S2 Figure S2. The atomic structures and ELFs for (B 12 )(CCC) shearing along the (01Ŭ11)/<101Ŭ1> slip system.