High Temperature Decomposition of Methane on Quartz Pellets
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Synthesis of Li2s-Carbon Cathode Materials Via Carbothermic Reduction of Li2so4
ORIGINAL RESEARCH published: 04 June 2019 doi: 10.3389/fenrg.2019.00053 Synthesis of Li2S-Carbon Cathode Materials via Carbothermic Reduction of Li2SO4 Jiayan Shi 1, Jian Zhang 2, Yifan Zhao 2, Zheng Yan 1, Noam Hart 1 and Juchen Guo 1,2* 1 Department of Chemical and Environmental Engineering, University of California, Riverside, Riverside, CA, United States, 2 Materials Science and Engineering Program, University of California, Riverside, Riverside, CA, United States Pre-lithiated sulfur materials are promising cathode for lithium-sulfur batteries. The synthesis of lithium sulfide-carbon (Li2S-C) composite by carbothermic reduction of lithium sulfate (Li2SO4) is investigated in this study. The relationship between reaction temperature and the consumption of carbon in the carbothermic reduction of Li2SO4 is first investigated to precisely control the carbon content in the resultant Li2S-C composites. To understand the relationship between the material structure and the electrochemical properties, Li2S-C composites with the same carbon content are subsequently synthesized by controlling the mass ratio of Li2SO4/carbon and the reaction temperature. Systematic electrochemical analyses and microscopic characterizations Edited by: demonstrate that the size of the Li S particles dispersed in the carbon matrix is the key Wen Liu, 2 Beijing University of Chemical parameter determining the electrochemical performance. A reversible capacity of 600 Technology, China −1 mAh g is achieved under lean electrolyte condition with high Li2S areal loading. -

Carbothermic Reduction Kinetics of Ilmenite Concentrates Catalyzed by Sodium Silicate and Microwave- Absorbing Characteristics O
Available on line at Association of the Chemical Engineers of Serbia AChE www.ache.org.rs/CICEQ Chemical Industry & Chemical Engineering Quarterly 19 (3) 423−433 (2013) CI&CEQ WEI LI1,2 CARBOTHERMIC REDUCTION KINETICS OF JINHUI PENG1 1 ILMENITE CONCENTRATES CATALYZED BY SHENGHUI GUO SODIUM SILICATE AND MICROWAVE- LIBO ZHANG1 GUO CHEN1 ABSORBING CHARACTERISTICS OF HONGYING XIA1 REDUCTIVE PRODUCTS 1 Key Laboratory of Unconventional Carbothermic reduction kinetics of ilmenite concentrates catalyzed by sodium Metallurgy, Ministry of Education, silicate were investigated; the reduction degree of ilmenite concentrates Kunming University of Science and reduction reaction was determined as R = 4/7(16y + 56x)(ΔWΣ - f W)/(16y + Technology, Yunnan, China A-P 2Faculty of Science, Kunming + 56x + 112). The results show that the reaction activation energy of initial University of Science and stage and later stage is 36.45 and 135.14 kJ/mol, respectively. There is a great Technology, Yunnan, China change in the reduction rate at temperatures of 1100 and 1150 °C; the catal- ysis effect and change of reduction rate were evaluated by TG and DSC curves SCIENTIFIC PAPER of sodium silicate. Microwave-absorbing characteristics of reduction products were measured by the method of microwave cavity perturbation. It was found UDC 544.47/.478 that microwave absorbing characteristics of reduction products obtained at ° DOI 10.2298/CICEQ120421077L temperatures of 900, 1100 and 1150 C have significant differences. XRD characterization results explained the formation and accumulation of reduction product Fe, and pronounced changes of microwave absorbing characteristics due to the decrease of the content of ilmenite concentrates. Keywords: sodium silicate; ilmenite concentrates; catalytic reduction; kinetics; microwave absorbing characteristics. -

CARBOTHERMIC REDUCTION of MECHANICALLY ACTIVATED Nio-CARBON MIXTURE: NON-ISOTHERMAL KINETICS
J. Min. Metall. Sect. B-Metall. 54 (3) B (2018) 313 - 322 Journal of Mining and Metallurgy, Section B: Metallurgy CARBOTHERMIC REDUCTION OF MECHANICALLY ACTIVATED NiO-CARBON MIXTURE: NON-ISOTHERMAL KINETICS S. Bakhshandeh, N. Setoudeh *, M. Ali Askari Zamani, A. Mohassel Yasouj University, Materials Engineering Department, Yasouj, Iran (Received 23 March 2018; accepted 10 December 2018) Abstract The effect of mechanical activation on the carbothermic reduction of nickel oxide was investigated. Mixtures of nickel oxide and activated carbon (99% carbon) were milled for different periods of time in a planetary ball mill. The unmilled mixture and milled samples were subjected to thermogravimetric analysis (TGA) under an argon atmosphere and their solid products of the reduction reaction were studied using XRD experiments. TGA showed that the reduction of NiO started at ~800 and ~720 in un-milled and one-hour milled samples respectively whilst after 25h of milling it decreased to about 430 . The kinetics parameters of carbothermic reduction were determined using non-isothermal method (Coats-Redfern Method) for un-milled and milled samples. The activation energy was determined to be about 222 kJ mol -1 for un-milled mixture whilst it was decreased to about 148 kJ mol -1 in 25-h milled sample. The decrease in the particle size/crystallite size of the℃ milled samp℃les resulted in a significant drop in the reaction temperature. ℃ Keywords : Ball milling; Carbothermic reduction; Nickel oxide; Non-isothermal kinetics; Thermo-gravimetric analysis (TGA). 1. Introduction solid product catalyzes the reaction rate [ 11 ]. It has been shown that mechanical activation by There have been many investigations on the milling reactants together with extended periods of carbothermic reduction of metal oxide in the literature time can intensify the reactions [ 16-19 ]. -

Carbothermic Production of Hexagonal Boron Nitride A
CARBOTHERMIC PRODUCTION OF HEXAGONAL BORON NITRIDE A THESIS SUBMITTED TO THE GRADUATE SCHOOL OF NATURAL AND APPLIED SCIENCES OF MIDDLE EAST TECHNICAL UNIVERSITY BY HASAN ERDEM ÇAMURLU IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY IN METALLURGICAL AND MATERIALS ENGINEERING NOVEMBER 2006 Approval of the Graduate School of Natural and Applied Sciences Prof. Dr. Canan Özgen Director I certify that this thesis satisfies all the requirements as a thesis for the degree of Doctor of Philosophy. Prof. Dr. Tayfur Öztürk Head of Department This is to certify that we have read this thesis and that in our opinion it is fully adequate, in scope and quality, as a thesis and for the degree of Doctor of Philosophy. Prof. Dr. Yavuz A. Topkaya Prof. Dr. Naci Sevinç Co-Supervisor Supervisor Examining Committee Members Prof. Dr Ahmet Geveci (METU, METE) Prof. Dr. Naci Sevinç (METU, METE) Prof. Dr. Yavuz A. Topkaya (METU, METE) Prof. Dr. Önder Özbelge (METU, CHE) Assoc. Prof. Dr. Nuri Durlu (ETÜ, ME) I hereby declare that all information in this document has been obtained and presented in accordance with academic rules and ethical conduct. I also declare that, as required by these rules and conduct, I have fully cited and referenced all material and results that are not original to this work. Name, Last name: Hasan Erdem Çamurlu Signature : ABSTRACT CARBOTHERMIC PRODUCTION OF HEXAGONAL BORON NITRIDE Çamurlu, Hasan Erdem Ph.D., Department of Metallurgical and Materials Engineering Supervisor : Prof. Dr. Naci Sevinç Co-Supervisor: Prof. Dr. Yavuz A. Topkaya October 2006, 124 pages Formation of hexagonal boron nitride (h-BN) by carbothermic reduction of B2O3 under nitrogen atmosphere at 1500oC was investigated. -
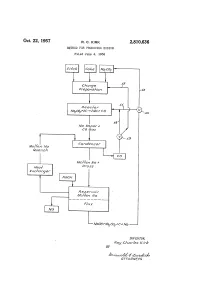
Wooh+Wacof C. Ma
Oct. 22, 1957 R. C. KRK 2,810,636 METHOD FOR PRODUCING SODIUM Filed June 4, 1956 Charge tAeac for Waco, 7-2C-2Wa CO Wa Vooor 7 CO Gas Mo/fern Wa Gornoeraser Guerch Mo/fer? Wa 7. Aea/ O/ro SS Af xcharger Aeser/or Mo/fer? Wa WoOh+WaCOf c. Ma INVENTOR. - Aoy CAar/es kirA BY A77OAAWEYS 2,810,636 United States Patent Office Patented Oct. 22, 1957 1. 2 Some dross is inevitably formed during this condensa tion operation and is separated from the molten sodium 2,810,636 by fluxing with sodium hydroxide. It has been discovered that the carbon monoxide which METHOD FOR PRODUCING SODUM is obtained as a by-product from the reaction of carbon with sodium carbonate will react with the sodium hy Roy.Chemical Charles Company,Kirk, Midland, Midland, Michassignor Mich., a corporation to The Dow of droxide in the flux at temperatures of at least 318 C. Delaware (at which temperature the sodium hydroxide is fluid or molten) to produce sodium carbonate. The thus treated Application June 4, 1956, Serial No. 589,198 0 flux may then be fed into the reactor with added carbon 7 Claims. (C. 75-66) to produce more sodium, since the higher melting sodium carbonate does not attack the graphite lining of the re actor. In this manner a cyclical process is obtained which recovers almost 100 percent of the sodium values present. This invention relates to a process for producing So 5 Since the sodium values in the dross are almost entirely dium and is particularly directed to an improved cyclical recovered, economical modifications of the sodium car method of making sodium by the carbothermic reduction bonate reduction process which previously could not be of sodium carbonate. -

Carbothermic Reduction of Alumina with Carbon in Vacuum
J. Cent. South Univ. (2012) 19: 1813−1816 DOI: 10.1007/s1177101212130 Carbothermic reduction of alumina with carbon in vacuum YU Qingchun(郁青春), YUAN Haibin(袁海滨), ZHU Fulong(朱富龙), ZHANG Han(张晗), WANG Chen(王辰), LIU Dachun(刘大春), YANG Bin(杨斌) National Engineering Laboratory of Vacuum Metallurgy, Key Laboratory of Breeding Base of Complex Nonferrous Metal Resources Clear Utilization in Yunnan Province, State Key Laboratory of Nonferrous Metal Vacuum Metallurgy, Kunming University of Science and Technology, Kunming 650093, China © Central South University Press and SpringerVerlag Berlin Heidelberg 2012 Abstract: Carbothermic reduction alumina in vacuum was conducted, and the products were analysed by means of XRD and gas chromatography. Thermodynamic analysis shows that in vacuum the initial carbothermic reduction reaction temperature reduces compared with that under normal pressure, and the preferential order of products is Al4O4C, Al4C3, Al2OC, Al2O and Al. Experiment results show that the carbothermic reduction products of alumina are Al4O4C and Al4C3, and neither Al2OC, Al2O or Al was found. During the carbothermic reduction process, the reaction rate of Al2O3 and carbon decreases gradually with increasing time. Meanwhile, lower system pressure or higher temperature is beneficial to the carbothermic reduction of alumina process. Al4O4C is firstly formed in the carbothermic reaction, and then Al4C3 is formed in lower system pressure or at higher temperature. Key words: alumina; carbothermic reduction; vacuum; aluminum the formation of aluminum carbide and oxycarbide 1 Introduction byproducts complicates the carbothermic reduction and chlorination process. The thermodynamic study of Aluminum is currently produced industrially via the chemical reactions provides a basic understanding of the HallHe´roult process by dissolving Al2O3 in fused process prior to designing suitable reaction experiments, NaFAlF3 (cryolite) followed by direct current and provides a useful guideline for the selection of electrolysis. -

Semi-Empirical Software for the Aluminothermic and Carbothermic Reactions
Association of Metallurgical Engineers of Serbia Scientific paper AMES UDC: 544.653.2:004.41 SEMI-EMPIRICAL SOFTWARE FOR THE ALUMINOTHERMIC AND CARBOTHERMIC REACTIONS Milorad Gavrilovski1, Vaso Manojlović2*, Željko Kamberović1, Marija Korać1, Miroslav Sokić2 1 Faculty of technology and metallurgy, Karnegijeva 4, University of Belgrade, Serbia 2 Institute for technology of nuclear and other mineral raw materials, Franše d’ Eperea 86, Belgrade, Serbia Received 23.06.2014 Accepted 03.09.2014 Abstract Understanding the reaction thermochemistry as well as formatting the empirical data about element distribution in gas-metal-slag phases is essential for creating a good model for aluminothermic and carbothermic reaction. In this paper modeling of material and energy balance of these reactions is described with the algorithm. The software, based on this model is basically made for production of high purity ferro alloys through aluminothermic process and then extended for some carbothermic process. Model validation is demonstrated with production of FeTi, FeW, FeB and FeMo in aluminothermic and reduction of mill scale, pyrite cinders and magnetite fines in carbothermic process. Introduction Aluminothermic reactions are thermite reactions in witch aluminum metal reacts with some metal oxide reducing it to the pure metal. These reactions are highly exothermic and self-propagating so only initiation is needed to complete the reaction. However, energy of reaction depends on various parameters and it its adjustment can be done by designing of thermite mixture. Aluminothermic reaction has found many applications in production of metals and alloys, welding and coating [1, 2]. In literature there is a lot of data on production, kinetics of reaction, preparation of thermite mixture, effects of additions on metal yield etc [1, 2]. -

Metallurgical Grade Silicon (MG-Si), Via Carbothermic Reduction
Engineering Impurity Behavior on the Micron-Scale in Metallurgical-Grade Silicon Production By Sarah Bernardis B.S./M.S. Physics Università degli Studi di Padova, 2005 M.S. Materials Science and Engineering Massachusetts Institute of Technology, 2008 Submitted to the Department of Materials Science and Engineering in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Materials Science and Engineering at the Massachusetts Institute of Technology February 2012 © 2012 Massachusetts Institute of Technology. All rights reserved. Signature of Author:_____________________________________________________________ Department of Materials Science and Engineering Certified by:___________________________________________________________________ Kenneth C. Russell Professor Emeritus of Metallurgy and Nuclear Engineering Thesis Supervisor Accepted by: __________________________________________________________________ Christopher Schuh Chair, Departmental Committee on Graduate Students - 2 - Engineering Impurity Behavior on the Micron-Scale in Metallurgical-Grade Silicon Production By Sarah Bernardis Submitted to the Department of Materials Science and Engineering on October 15th, 2012 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Materials Science and Engineering Abstract Impurities are detrimental to silicon-based solar cells. A deeper understanding of their evolution, microscopic distributions, and oxidation states throughout the refining processes may enable the discovery of novel refining techniques. Using synchrotron-based microprobe techniques and bulk chemical analyses, we investigate Fe, Ti, and Ca starting from silicon- and carbon bearing raw feedstock materials to metallurgical grade silicon (MG-Si), via carbothermic reduction. Before reduction, impurities are present in distinct micron- or sub-micron-sized minerals, frequently located at structural defects in Si-bearing compounds. Chemical states vary, they are generally oxidized (e.g., Fe2+, Fe3+). -

Technological Gaps Inhibiting the Exploitation of Crms Secondary Resources
SCRREEN Coordination and Support Action (CSA) This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No 730227. Start date : 2016-11-01 Duration : 38 Months www.scrreen.eu Technological gaps inhibiting the exploitation of CRMs secondary resources Authors : Mr. Stéphane BOURG (CEA) SCRREEN - D6.2 - Issued on 2019-11-07 09:13:37 by CEA SCRREEN - D6.2 - Issued on 2019-11-07 09:13:37 by CEA SCRREEN - Contract Number: 730227 Project officer: Jonas Hedberg Document title Technological gaps inhibiting the exploitation of CRMs secondary resources Author(s) Mr. Stéphane BOURG Number of pages 257 Document type Deliverable Work Package WP6 Document number D6.2 Issued by CEA Date of completion 2019-11-07 09:13:37 Dissemination level Public Summary Technological gaps inhibiting the exploitation of CRMs secondary resources Approval Date By 2019-11-21 12:54:45 Mrs. Maria TAXIARCHOU (LabMet, NTUA) 2019-11-21 14:40:17 Mr. Stéphane BOURG (CEA) SCRREEN - D6.2 - Issued on 2019-11-07 09:13:37 by CEA SCRREEN This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No 730227. Start date: 2016-11-01 Duration: 37 Months DELIVERABLE 6.2: TECHNOLOGICAL GAPS INHIBITING THE EXPLOITATION OF CRM SECONDARY RESOURCES AUTHOR(S): AMPHOS21, BRGM, CEA, CHALMERS, IDENER, SWERIM, NTUA Writing coordinated by S. Bourg, CEA DATE OF FIRST SUBMISSION: 30/04/2019 CONTENT Figures And Tables ............................................................................................................................................. -

Kinetic Model for the Uniform Conversion of Self Reducing Iron Oxide and Carbon Briquettes
ISIJ International, Vol. 43 (2003), No. 8, pp. 1136–1142 Kinetic Model for the Uniform Conversion of Self Reducing Iron Oxide and Carbon Briquettes Jeremy MOON and Veena SAHAJWALLA School of Materials Science and Engineering, University of New South Wales, Sydney 2052 NSW, Australia. (Received on September 2, 2002; accepted in final form on February 5, 2003 ) A kinetic model has been developed to describe the uniform conversion of a self reducing mixture of iron oxide and carbon. The model takes into account the reaction kinetics of both the iron oxide reduction and carbon oxidation. The model is validated with experimental data. Rate constants are compared with those in the literature. The combination of existing reaction analysis techniques coupled with the model developed has shown that for the experimental conditions used here, the Boudouard reaction controls the self reduction kinetics. KEY WORDS: mathematical modelling; kinetics; uniform conversion model; self-reduction; iron oxide; car- bon; reduction; oxidation. that for practical situations, self reducing mixtures undergo 1. Introduction indirect reduction (solid iron oxide–gaseous intermediary– Extensive research regarding the self-reducing mixture of solid carbon). These reactions are outlined in Reaction 1 to iron oxide and carbonaceous materials has been report- Reaction 4. ed.1–5) Iron oxide fines are generated at every stage of iron Reduction oxide processing, leading to either a loss of the iron re- source or the burden of higher utilisation costs. Being able Reaction 1: 3Fe O ϩCO→Fe O ϩCO to utilise, economically, such iron oxide fines has the poten- 2 3 3 4 2 tial to provide significant benefits to the iron industry. -

STUDY of CARBON AEROGEL SUPPORTED Fe CATALYSTS for BIOMASS GASIFICATION GAS CLEANING
UNIVERSIDAD DE CONCEPCIÓN DEPARTAMENTO DE INGENIERÍA QUÍMICA STUDY OF CARBON AEROGEL SUPPORTED Fe CATALYSTS FOR BIOMASS GASIFICATION GAS CLEANING POR OSCAR GÓMEZ CÁPIRO Tesis presentada a la Facultad de Ingeniería de la Universidad de Concepción para optar el grado de Doctor en Ciencias de la Ingeniería con mención en Ingeniería Química Tutor: Prof. Romel Mario Jiménez Concepción, PhD. Departamento de Ingeniería Química Universidad de Concepción Co-tutor: Prof. Luis Ernesto Arteaga Pérez, PhD. Departamento de Ingeniería en Madera Universidad del Bío-Bío Concepción, Chile. Junio de 2020 Se autoriza la reporducción total o parcial , con fines académicos , por cualquier medio o procedimiento, incluyendo la cita bibliográfica del documento. Advisor: Prof. Romel Mario Jiménez Concepción, PhD. Chemical Engineering Department University of Concepción Co-advisor: Prof. Luis Ernesto Arteaga Pérez, PhD. Department of Wood Engineering University of Bío-Bío Examination Prof. Ximena García Carmona, PhD. Committee Chemical Engineering Department University of Concepción Prof. Fernando Mariño, PhD. ITHES University of Buenos Aires; CONICET i Dedicatoria Dedicatoria A mi abuela Dora, Doña Cedora del Rosario Muñoz y Anoceto… ii Agradeciemientos Agradecimientos Quisiera aprovechar para agradecer a algunas de las personas que han hecho posible que se realizara el trabajo que se detalla más adelante. Comenzaré por dar gracias al profesor Luis Ernesto Arteaga Pérez que no solo me guío académicamente, sino que me ofreció apoyo en todo, abriendo su casa y su familia para recibirme en Chile, por eso debo agradecer también a su esposa Yannay Casas Ledón. Quiero dar gracias al profesor Romel Mario Jiménez Concepción quien guió mi trabajo académico y mi formación como doctor. -

Tore Meberg.Pdf (14.85Mb)
Redesign of Utility Robot for Carbothermic Test Reactor Tore Meberg Supervisors Cecilie Ødegård John Fors This master’s thesis is carried out as a part of the education at the University of Agder and is therefore approved as a part of this education. However, this does not imply that the University answers for the methods that are used or the conclusions that are drawn. University of Agder, 2015 Faculty of Engineering and Science Department of Engineering Sciences Abstract Production of aluminum based on a carbothermic reaction is an exciting field of research. It could potentially reduce the energy consumption of aluminum production drastically. Alcoa Norway Carbothermic has been involved in the field of carbothermic aluminum research since 1998. As a part of development of their newest carbothermic test reactor, the Hot Bowl reactor, Alcoa Norway Carbothermic has proposed a thesis regarding redesign of a machine used to lower different types of measuring equipment down into the reactor, called the utility robot. This thesis describe the process leading up to a new utility robot solution. The new utility robot was designed in SolidWorks and dimensioned using calculations and FEM analyses in Abaqus. Technical drawings were composed for all fully developed components. These components were then issued for construction, and are currently being machined. Pneumatic and hydraulic cylinders, linear rails, bearings and other components were selected and ordered. The final result is a nearly complete utility robot that fulfill the requirements and design specifications specified by Alcoa. The new solution is more automatic and is less labor intensive to operate than the old solution.